Modlin Samuel J, Elghraoui Afif, Gunasekaran Deepika, Zlotnicki Alyssa M, Dillon Nicholas A, Dhillon Nermeeta, Kuo Norman, Robinhold Cassidy, Chan Carmela K, Baughn Anthony D, Valafar Faramarz
Laboratory for Pathogenesis of Clinical Drug Resistance and Persistence, San Diego State Universitygrid.263081.e, San Diego, California, USA.
Department of Microbiology and Immunology, University of Minnesotagrid.17635.36 Medical School, Minneapolis, Minnesota, USA.
mSystems. 2021 Dec 21;6(6):e0067321. doi: 10.1128/mSystems.00673-21. Epub 2021 Nov 2.
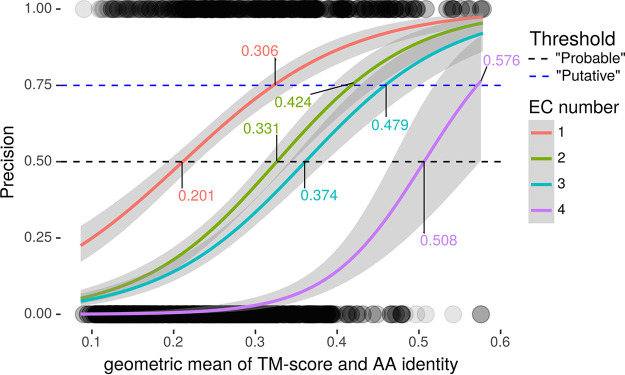
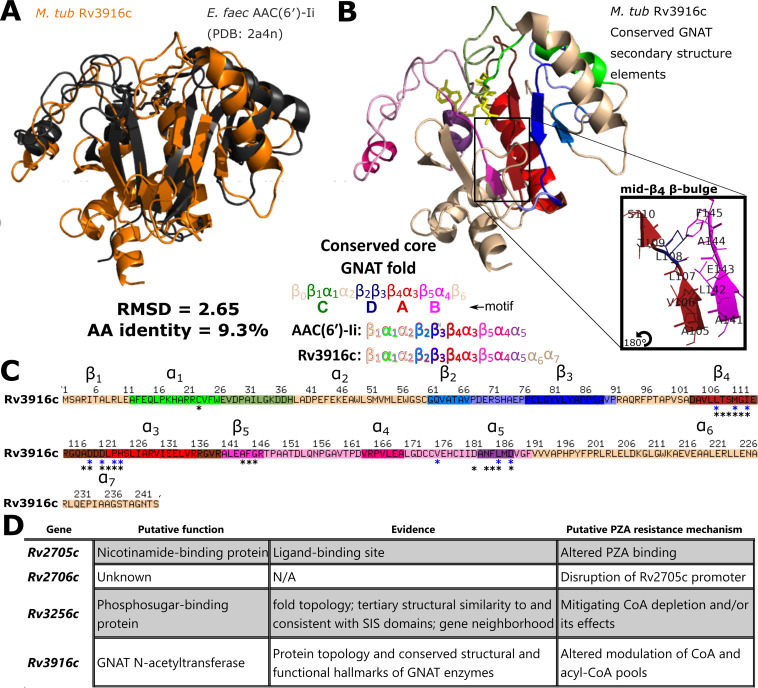
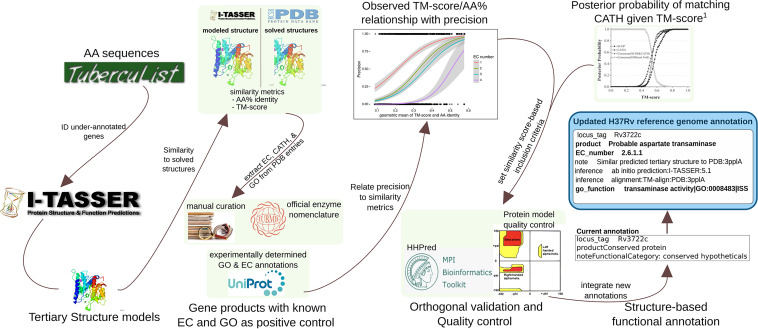
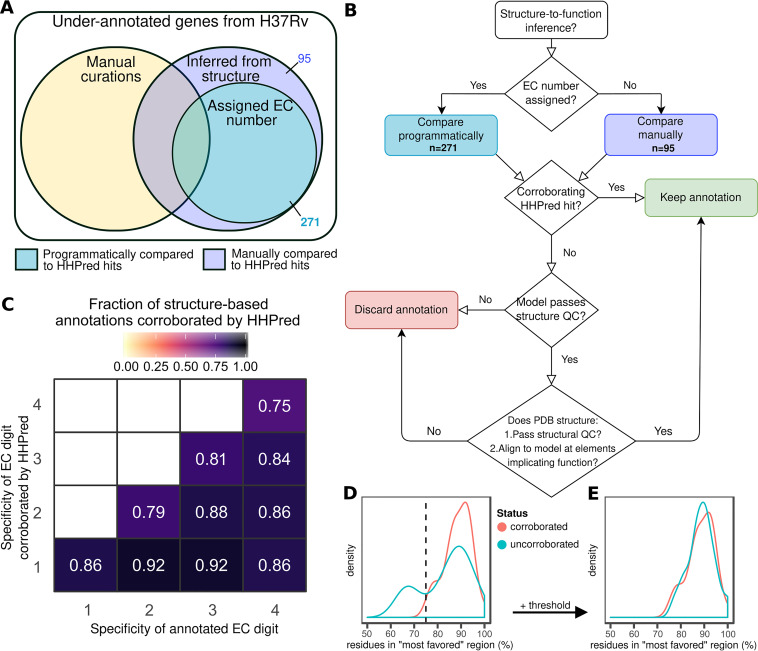
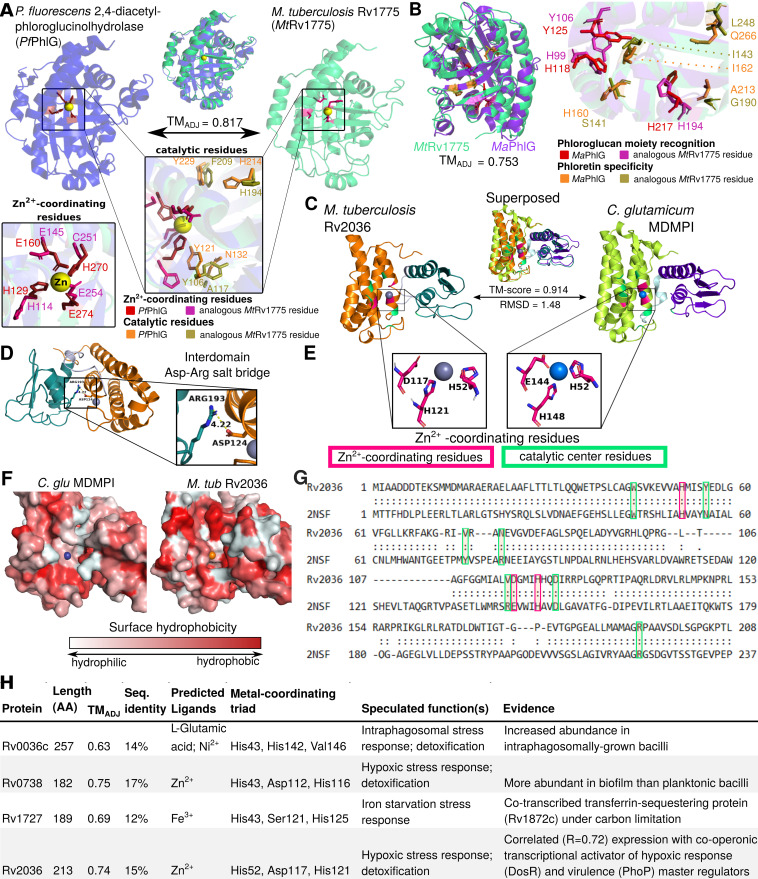
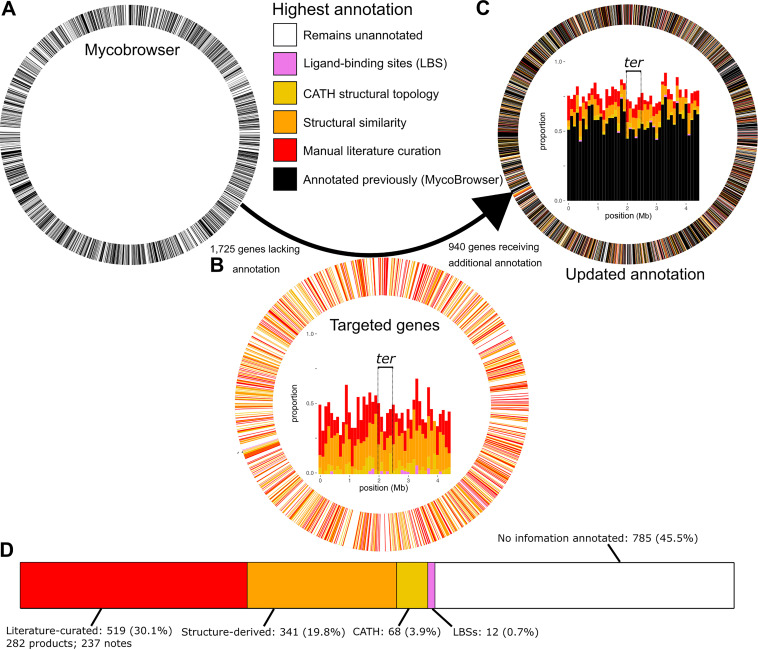
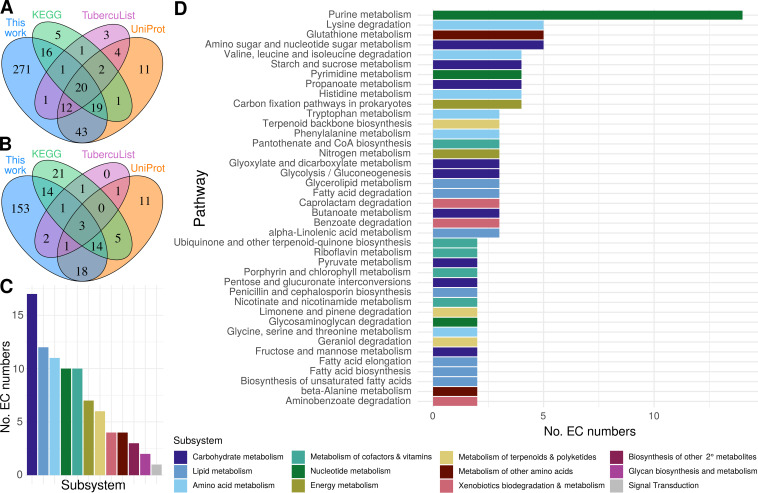
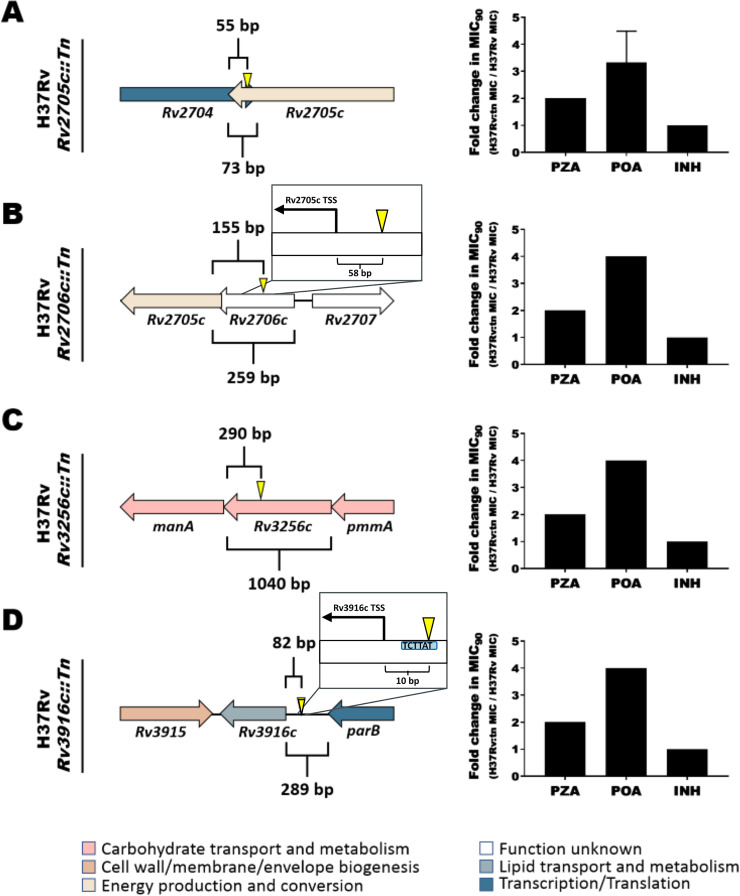
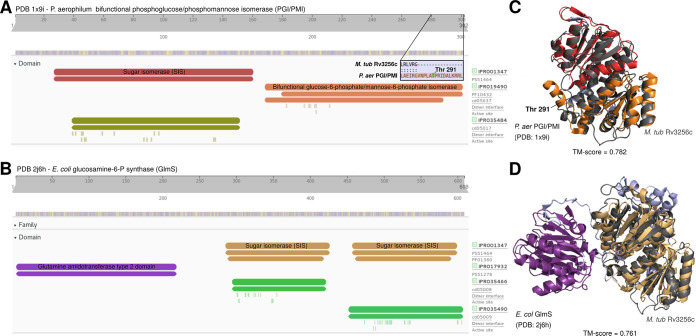
Accurate and timely functional genome annotation is essential for translating basic pathogen research into clinically impactful advances. Here, through literature curation and structure-function inference, we systematically update the functional genome annotation of Mycobacterium tuberculosis virulent type strain H37Rv. First, we systematically curated annotations for 589 genes from 662 publications, including 282 gene products absent from leading databases. Second, we modeled 1,711 underannotated proteins and developed a semiautomated pipeline that captured shared function between 400 protein models and structural matches of known function on Protein Data Bank, including drug efflux proteins, metabolic enzymes, and virulence factors. In aggregate, these structure- and literature-derived annotations update 940/1,725 underannotated H37Rv genes and generate hundreds of functional hypotheses. Retrospectively applying the annotation to a recent whole-genome transposon mutant screen provided missing function for 48% (13/27) of underannotated genes altering antibiotic efficacy and 33% (23/69) required for persistence during mouse tuberculosis (TB) infection. Prospective application of the protein models enabled us to functionally interpret novel laboratory generated pyrazinamide (PZA)-resistant mutants of unknown function, which implicated the emerging coenzyme A depletion model of PZA action in the mutants' PZA resistance. Our findings demonstrate the functional insight gained by integrating structural modeling and systematic literature curation, even for widely studied microorganisms. Functional annotations and protein structure models are available at https://tuberculosis.sdsu.edu/H37Rv in human- and machine-readable formats. Mycobacterium tuberculosis, the primary causative agent of tuberculosis, kills more humans than any other infectious bacterium. Yet 40% of its genome is functionally uncharacterized, leaving much about the genetic basis of its resistance to antibiotics, capacity to withstand host immunity, and basic metabolism yet undiscovered. Irregular literature curation for functional annotation contributes to this gap. We systematically curated functions from literature and structural similarity for over half of poorly characterized genes, expanding the functionally annotated Mycobacterium tuberculosis proteome. Applying this updated annotation to recent functional screens added functional information to dozens of clinically pertinent proteins described as having unknown function. Integrating the annotations with a prospective functional screen identified new mutants resistant to a first-line TB drug, supporting an emerging hypothesis for its mode of action. These improvements in functional interpretation of clinically informative studies underscore the translational value of this functional knowledge. Structure-derived annotations identify hundreds of high-confidence candidates for mechanisms of antibiotic resistance, virulence factors, and basic metabolism and other functions key in clinical and basic tuberculosis research. More broadly, they provide a systematic framework for improving prokaryotic reference annotations.
准确及时的功能基因组注释对于将基础病原体研究转化为具有临床影响力的进展至关重要。在此,通过文献整理和结构 - 功能推断,我们系统地更新了结核分枝杆菌强毒株H37Rv的功能基因组注释。首先,我们系统地整理了来自662篇出版物中589个基因的注释,其中包括主要数据库中缺失的282个基因产物。其次,我们对1711个注释不足的蛋白质进行建模,并开发了一个半自动流程,该流程捕捉了400个蛋白质模型与蛋白质数据库中已知功能的结构匹配之间的共同功能,包括药物外排蛋白、代谢酶和毒力因子。总体而言,这些基于结构和文献的注释更新了1725个注释不足的H37Rv基因中的940个,并产生了数百个功能假设。将该注释追溯应用于最近的全基因组转座子突变体筛选,为48%(13/27)注释不足且改变抗生素疗效的基因以及33%(23/69)在小鼠结核病(TB)感染期间持续存在所需的基因提供了缺失的功能。蛋白质模型的前瞻性应用使我们能够从功能上解释新产生的功能未知的吡嗪酰胺(PZA)耐药突变体,这暗示了PZA作用的新兴辅酶A消耗模型与突变体对PZA的耐药性有关。我们的研究结果表明,即使对于广泛研究的微生物,通过整合结构建模和系统的文献整理也能获得功能见解。功能注释和蛋白质结构模型可在https://tuberculosis.sdsu.edu/H37Rv以人类可读和机器可读格式获取。结核分枝杆菌是结核病的主要病原体,其致死人数超过任何其他传染性细菌。然而,其40%的基因组在功能上尚未表征,其抗生素耐药性、抵御宿主免疫的能力以及基本代谢的遗传基础仍有许多未被发现。功能注释的不规则文献整理导致了这一差距。我们系统地从文献和结构相似性中整理了超过一半特征不佳基因的功能,扩展了功能注释的结核分枝杆菌蛋白质组。将这种更新后的注释应用于最近的功能筛选,为数十个被描述为功能未知的临床相关蛋白质添加了功能信息。将这些注释与前瞻性功能筛选相结合,鉴定出对一线结核病药物耐药的新突变体,支持了其作用模式的新兴假设。这些对临床信息研究功能解释的改进强调了这种功能知识的转化价值。基于结构的注释识别出数百个关于抗生素耐药机制、毒力因子、基本代谢以及临床和基础结核病研究中其他关键功能的高可信度候选者。更广泛地说,它们为改进原核生物参考注释提供了一个系统框架。