CSE and IT Department; School of Electrical Engineering and Computer; Shiraz University, Shiraz, Iran.
Department of Medicinal Chemistry, School of Pharmacy, Shiraz University of Medical Sciences, Shiraz, Iran.
Comput Biol Med. 2021 Dec;139:104967. doi: 10.1016/j.compbiomed.2021.104967. Epub 2021 Oct 25.
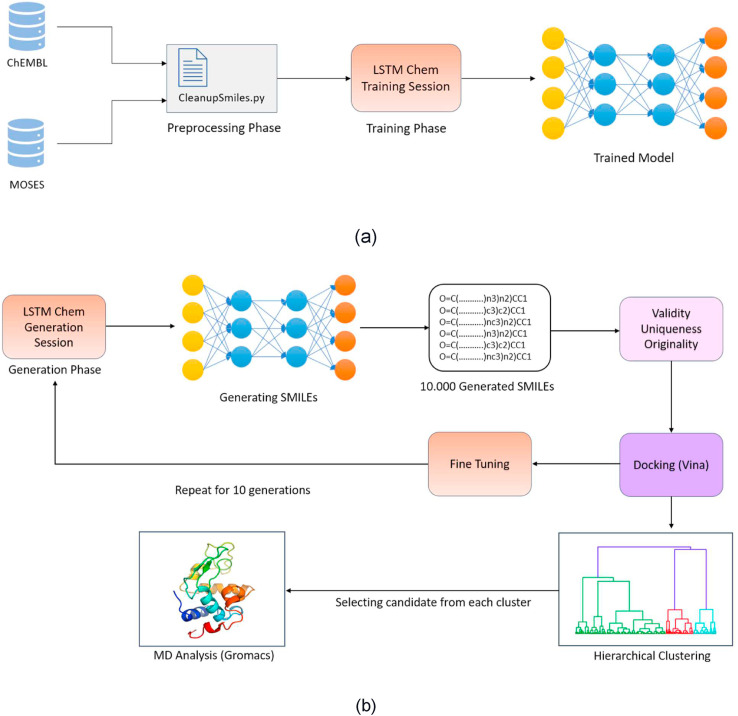
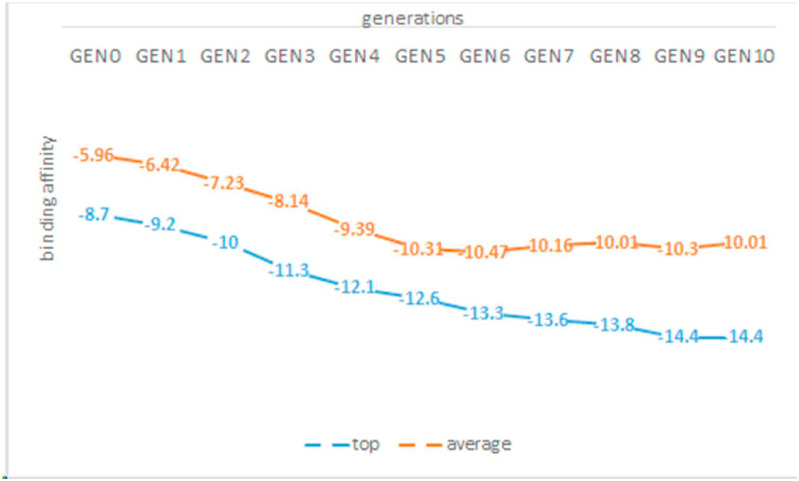
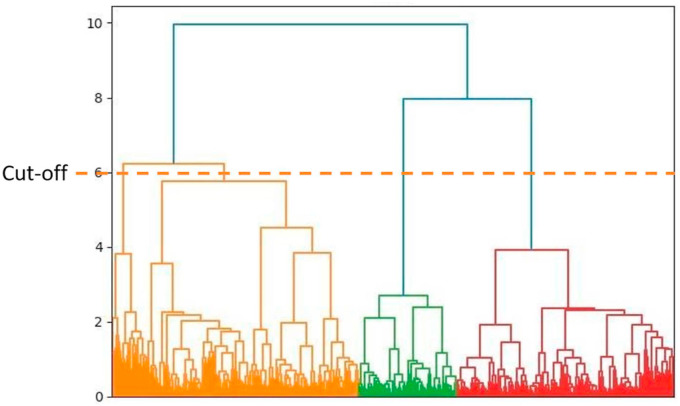
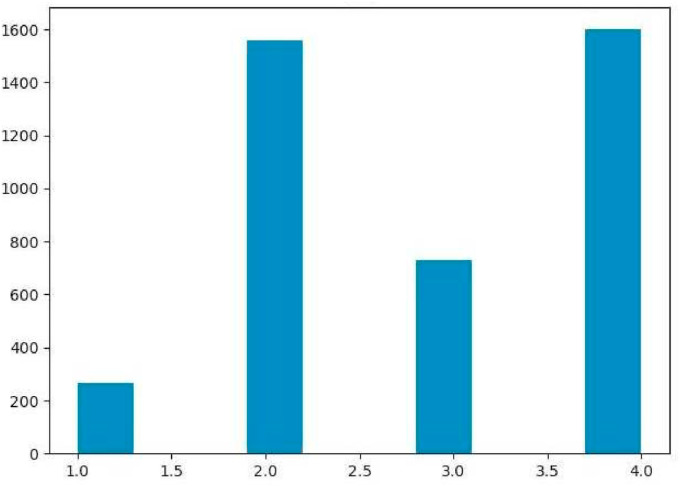
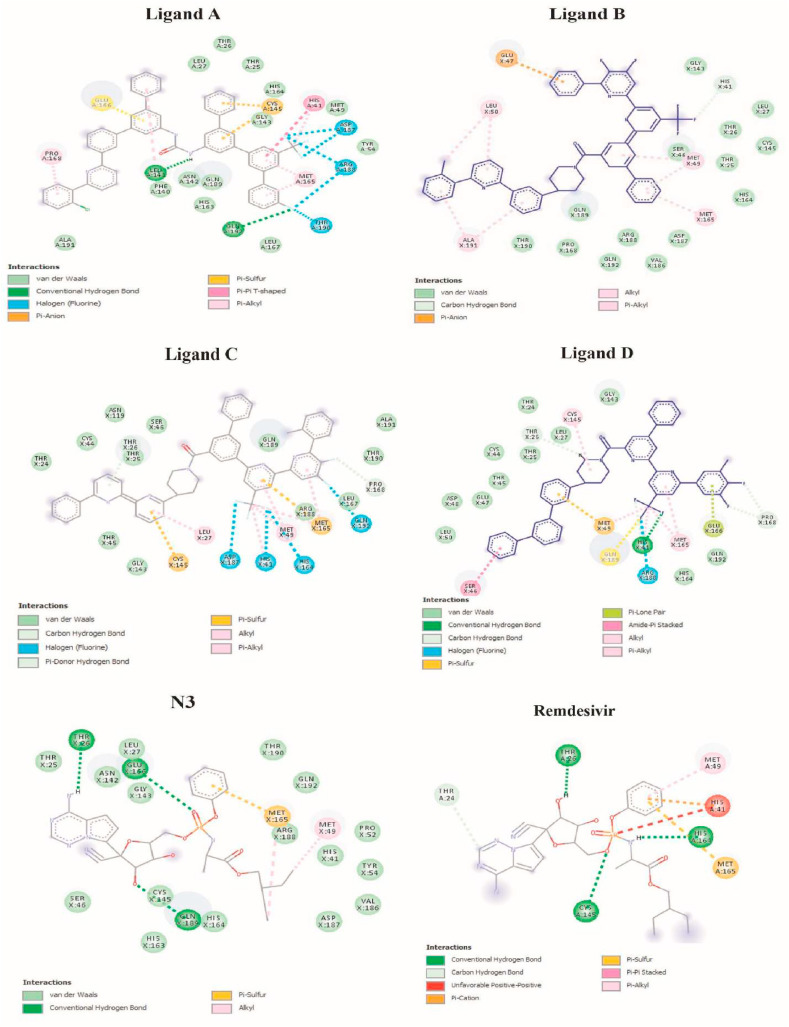
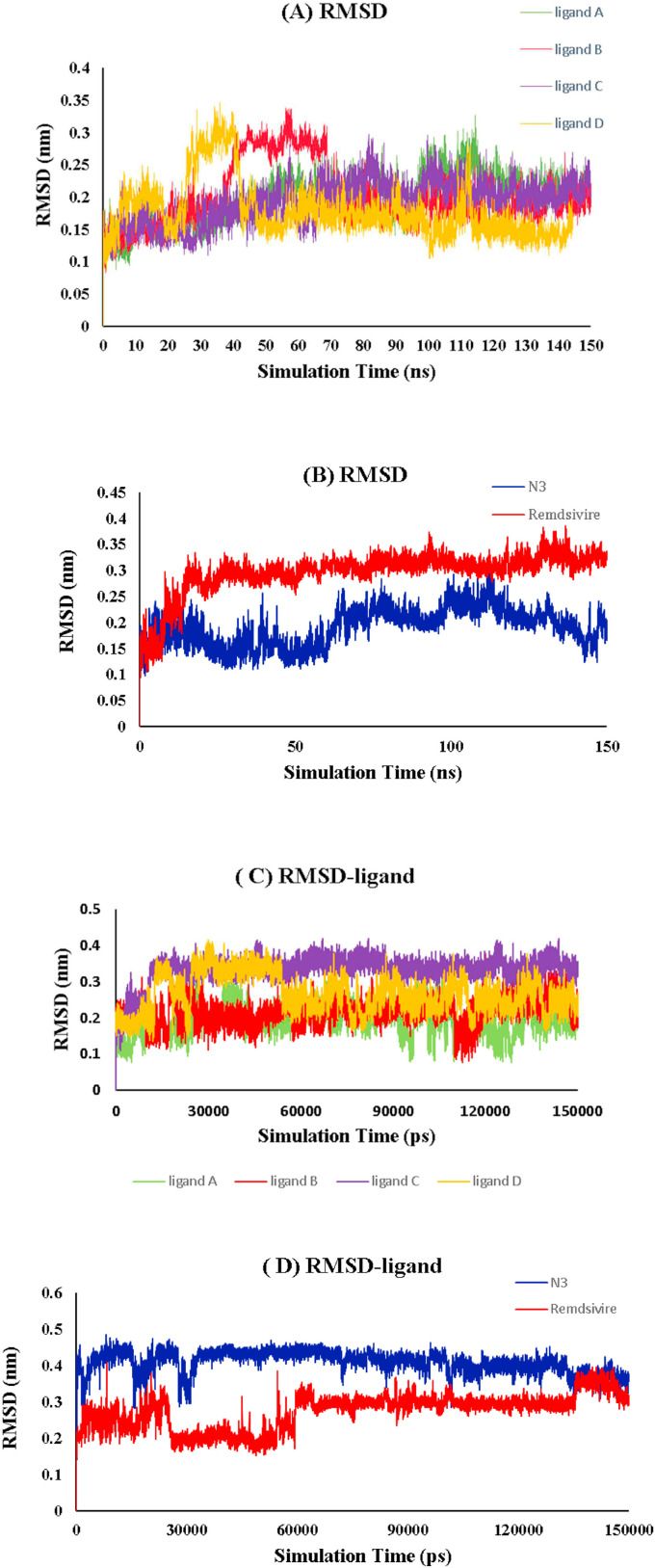
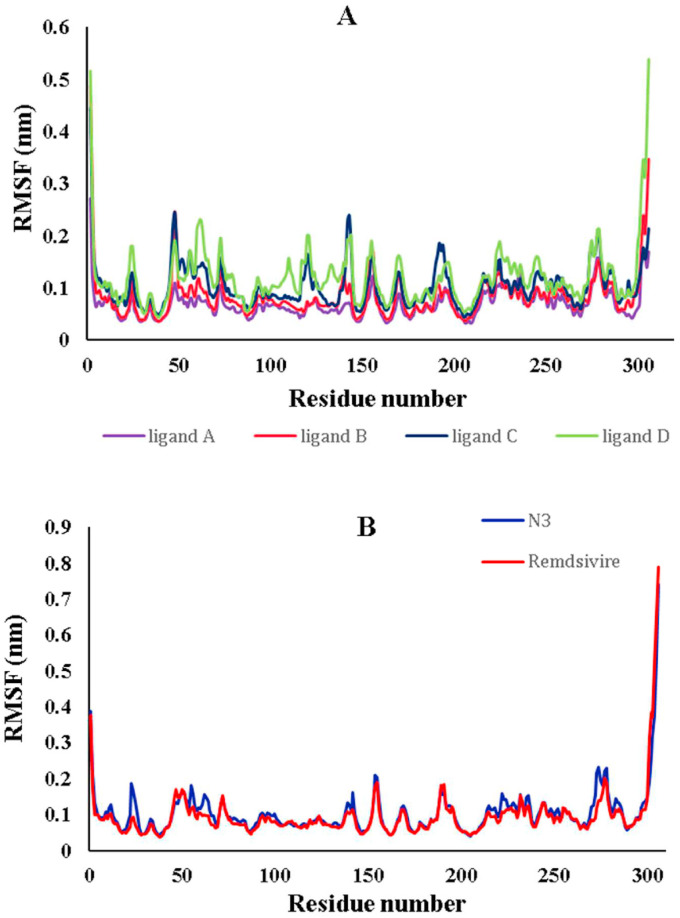
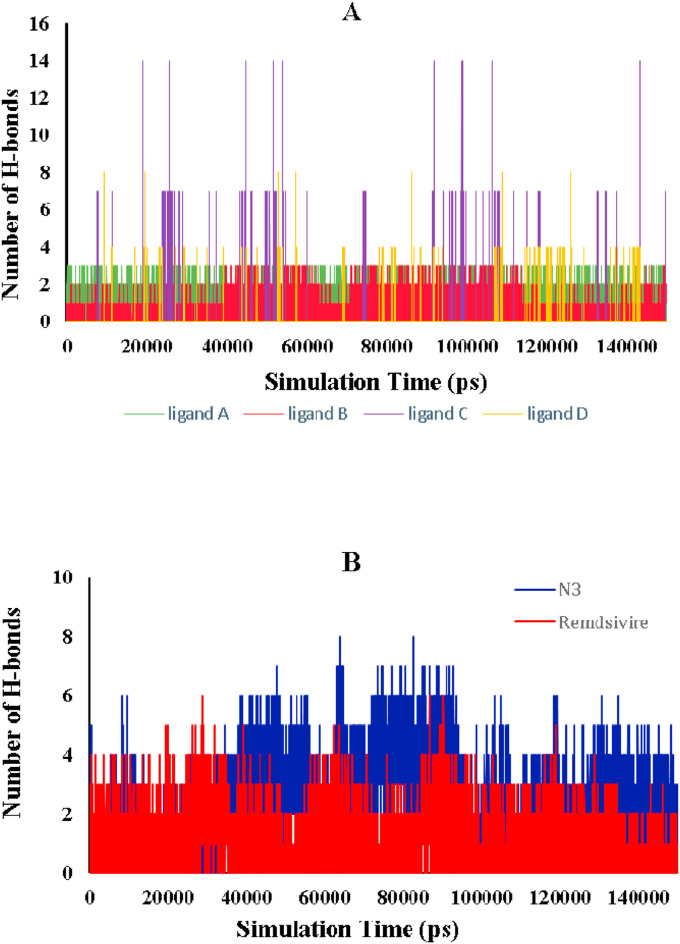
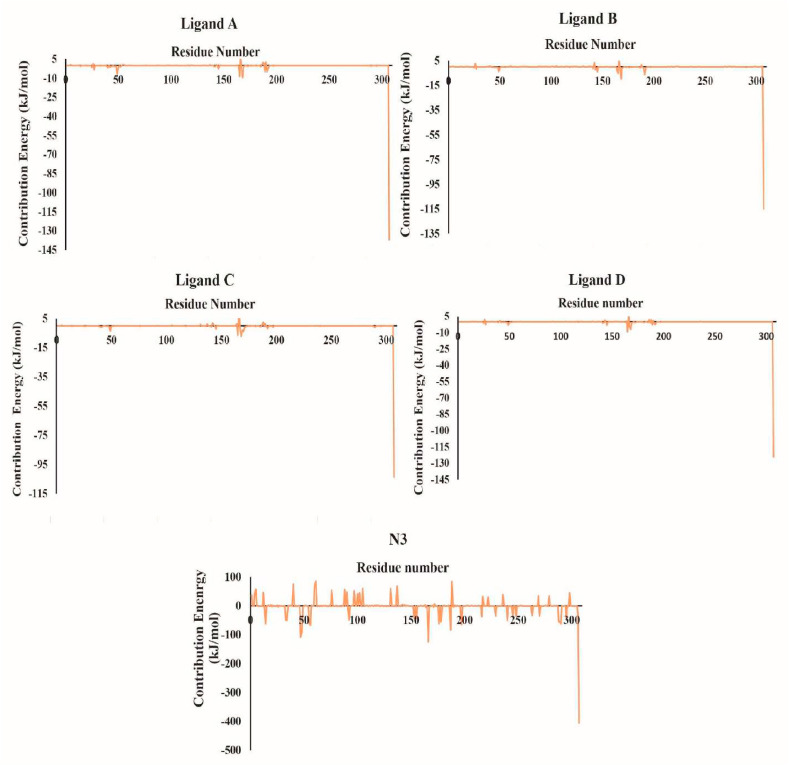
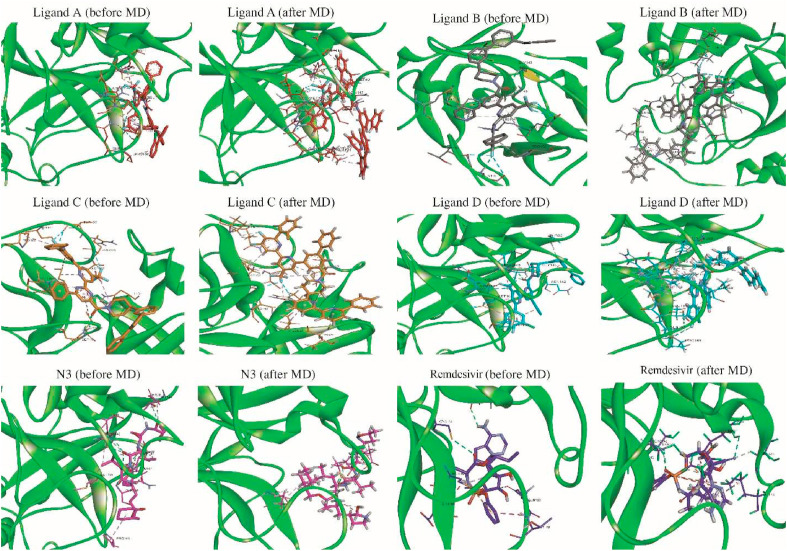
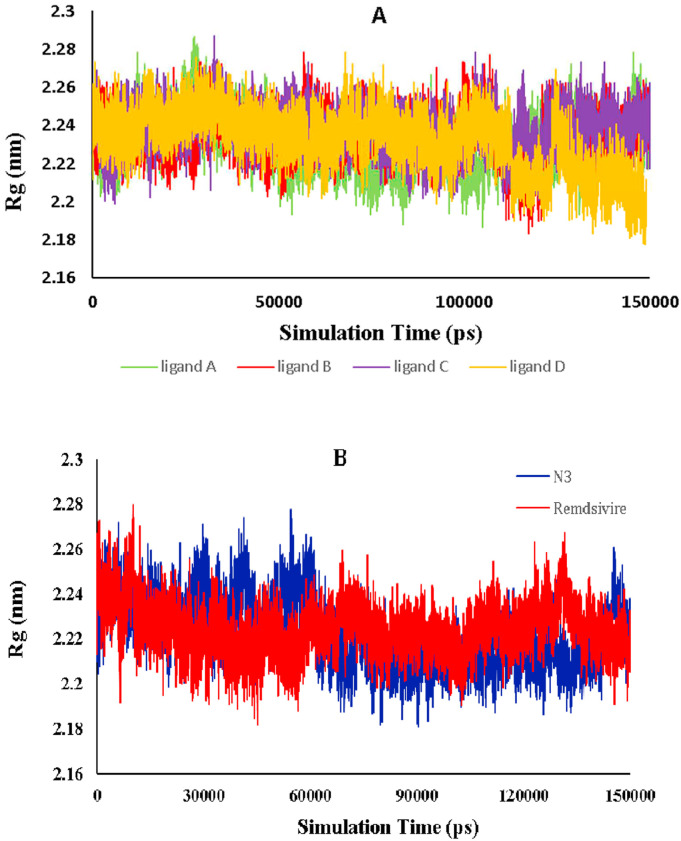
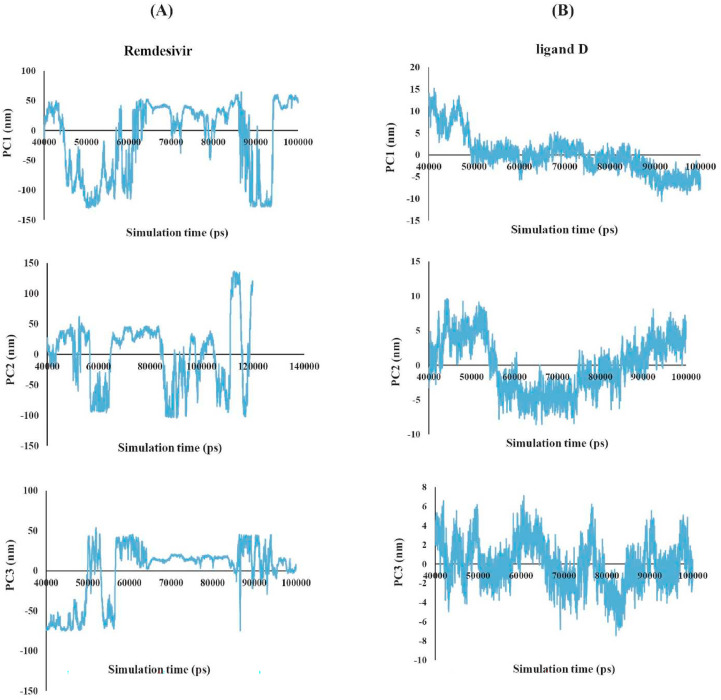
The main protease of SARS-CoV-2 is a critical target for the design and development of antiviral drugs. 2.5 M compounds were used in this study to train an LSTM generative network via transfer learning in order to identify the four best candidates capable of inhibiting the main proteases in SARS-CoV-2. The network was fine-tuned over ten generations, with each generation resulting in higher binding affinity scores. The binding affinities and interactions between the selected candidates and the SARS-CoV-2 main protease are predicted using a molecular docking simulation using AutoDock Vina. The compounds selected have a strong interaction with the key MET 165 and Cys145 residues. Molecular dynamics (MD) simulations were run for 150ns to validate the docking results on the top four ligands. Additionally, root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), and hydrogen bond analysis strongly support these findings. Furthermore, the MM-PBSA free energy calculations revealed that these chemical molecules have stable and favorable energies, resulting in a strong binding with Mpro's binding site. This study's extensive computational and statistical analyses indicate that the selected candidates may be used as potential inhibitors against the SARS-CoV-2 in-silico environment. However, additional in-vitro, in-vivo, and clinical trials are required to demonstrate their true efficacy.
SARS-CoV-2 的主要蛋白酶是设计和开发抗病毒药物的关键靶标。本研究使用 2.5M 化合物通过迁移学习训练 LSTM 生成网络,以鉴定四种最有能力抑制 SARS-CoV-2 主要蛋白酶的候选药物。该网络经过十代的微调,每一代都产生了更高的结合亲和力评分。使用 AutoDock Vina 进行分子对接模拟预测所选候选药物与 SARS-CoV-2 主要蛋白酶之间的结合亲和力和相互作用。所选化合物与关键 MET165 和 Cys145 残基具有强烈的相互作用。对前四个配体进行了 150ns 的分子动力学 (MD) 模拟,以验证对接结果。此外,均方根偏差 (RMSD)、均方根波动 (RMSF) 和氢键分析也强烈支持这些发现。此外,MM-PBSA 自由能计算表明,这些化学分子具有稳定且有利的能量,与 Mpro 的结合位点具有很强的结合能力。这项研究的广泛计算和统计分析表明,所选候选药物可能在 SARS-CoV-2 的计算机环境中用作潜在的抑制剂。然而,需要进行更多的体外、体内和临床试验来证明它们的真正疗效。