Agroscopegrid.417771.3, Method Development and Analytics & SIB Swiss Institute of Bioinformatics, Wädenswil, Switzerland.
Wadsworth Center, New York State Department of Health, Albany, New York, USA.
J Bacteriol. 2022 Jan 18;204(1):e0035321. doi: 10.1128/JB.00353-21. Epub 2021 Nov 8.
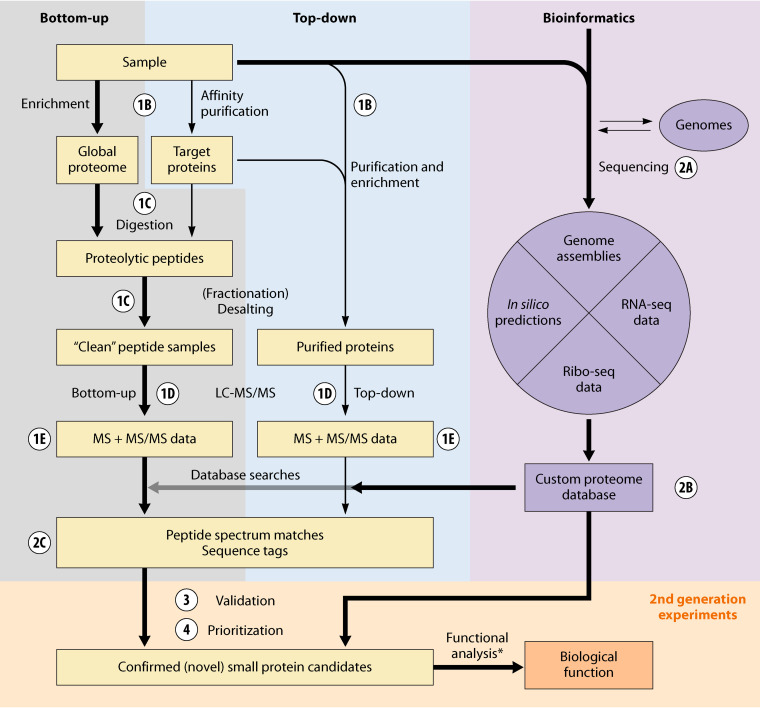
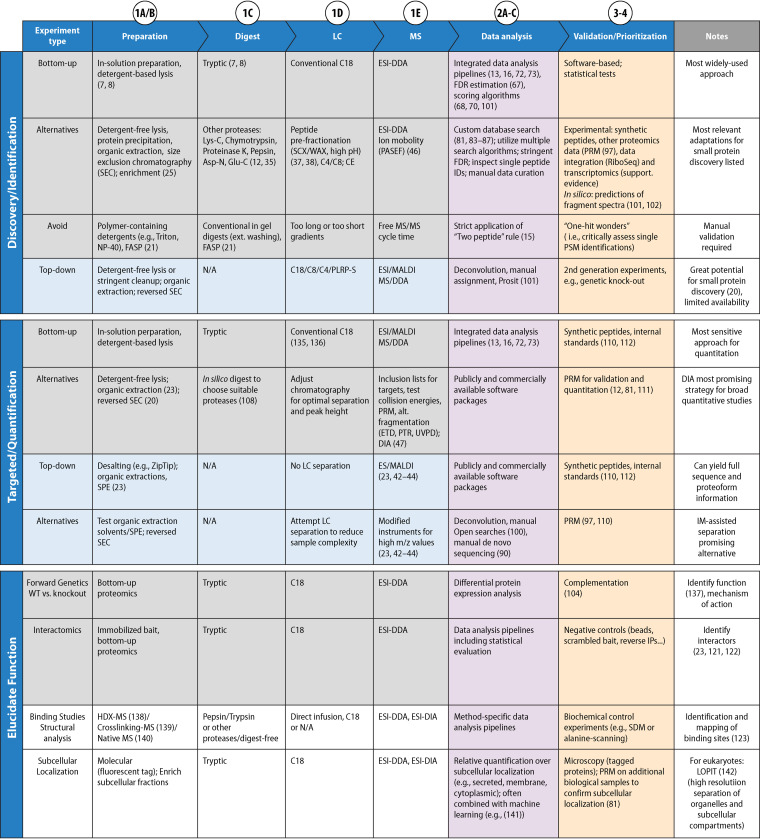
Small proteins of up to ∼50 amino acids are an abundant class of biomolecules across all domains of life. Yet due to the challenges inherent in their size, they are often missed in genome annotations, and are difficult to identify and characterize using standard experimental approaches. Consequently, we still know few small proteins even in well-studied prokaryotic model organisms. Mass spectrometry (MS) has great potential for the discovery, validation, and functional characterization of small proteins. However, standard MS approaches are poorly suited to the identification of both known and novel small proteins due to limitations at each step of a typical proteomics workflow, i.e., sample preparation, protease digestion, liquid chromatography, MS data acquisition, and data analysis. Here, we outline the major MS-based workflows and bioinformatic pipelines used for small protein discovery and validation. Special emphasis is placed on highlighting the adjustments required to improve detection and data quality for small proteins. We discuss both the unbiased detection of small proteins and the targeted analysis of small proteins of interest. Finally, we provide guidelines to prioritize novel small proteins, and an outlook on methods with particular potential to further improve comprehensive discovery and characterization of small proteins.
小蛋白是一类丰度很高的生物分子,其大小约为 50 个氨基酸。然而,由于其大小的固有挑战,它们在基因组注释中经常被遗漏,并且使用标准实验方法也难以识别和表征。因此,即使在研究良好的原核模式生物中,我们对小蛋白的了解仍然很少。质谱 (MS) 具有发现、验证和功能表征小蛋白的巨大潜力。然而,由于典型蛋白质组学工作流程的每个步骤都存在局限性,即样品制备、蛋白酶消化、液相色谱、MS 数据采集和数据分析,标准 MS 方法不太适合鉴定已知和新型小蛋白。在这里,我们概述了用于小蛋白发现和验证的主要基于 MS 的工作流程和生物信息学管道。特别强调了为提高小蛋白的检测和数据质量所需的调整。我们讨论了小蛋白的无偏检测和感兴趣的小蛋白的靶向分析。最后,我们提供了优先考虑新型小蛋白的指导方针,并展望了具有特别潜力进一步提高小蛋白综合发现和表征的方法。