The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
Texas Tech University Health Science Center, El Paso, TX, USA.
Mod Pathol. 2022 Apr;35(4):470-479. doi: 10.1038/s41379-021-00961-0. Epub 2021 Nov 13.
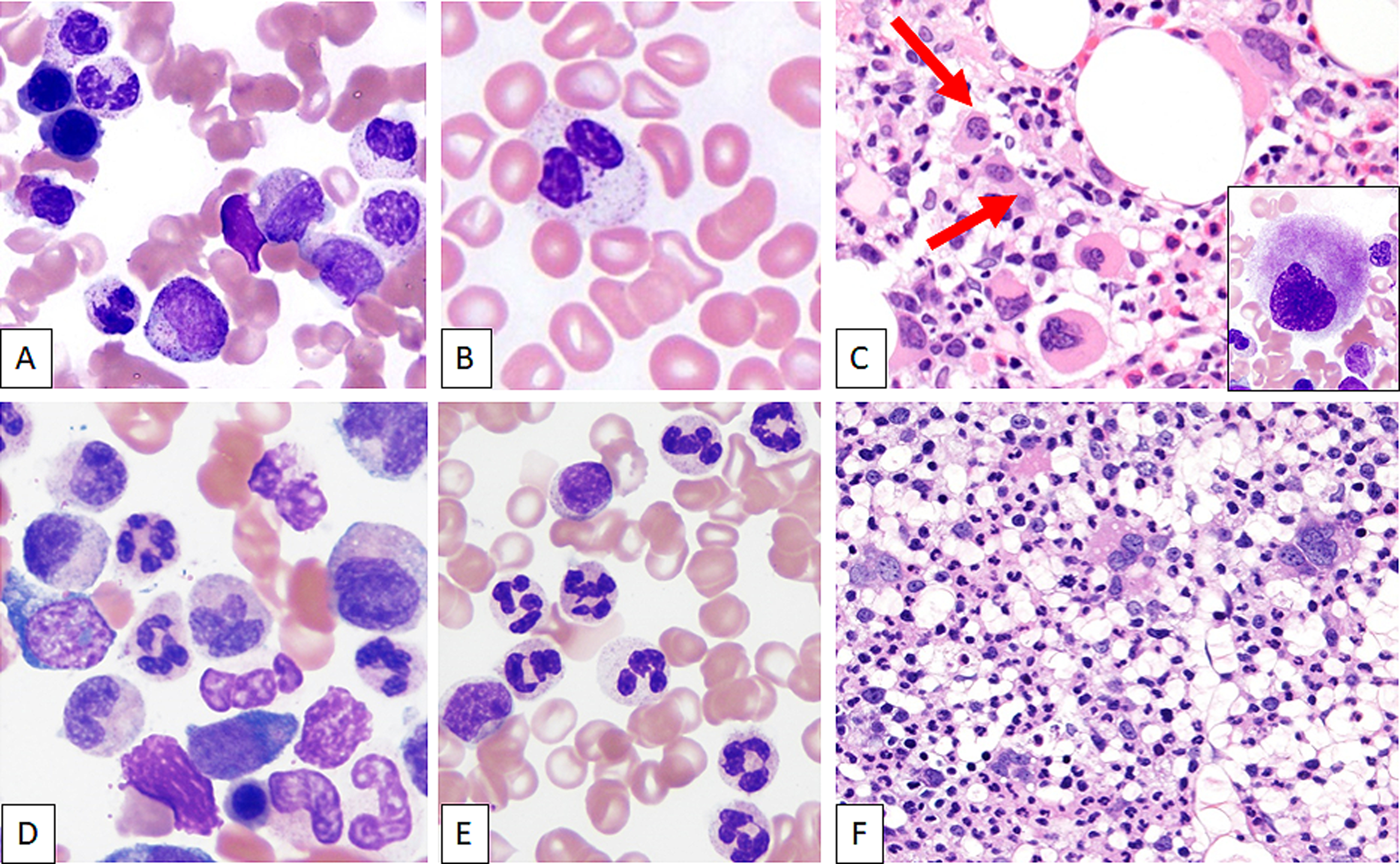
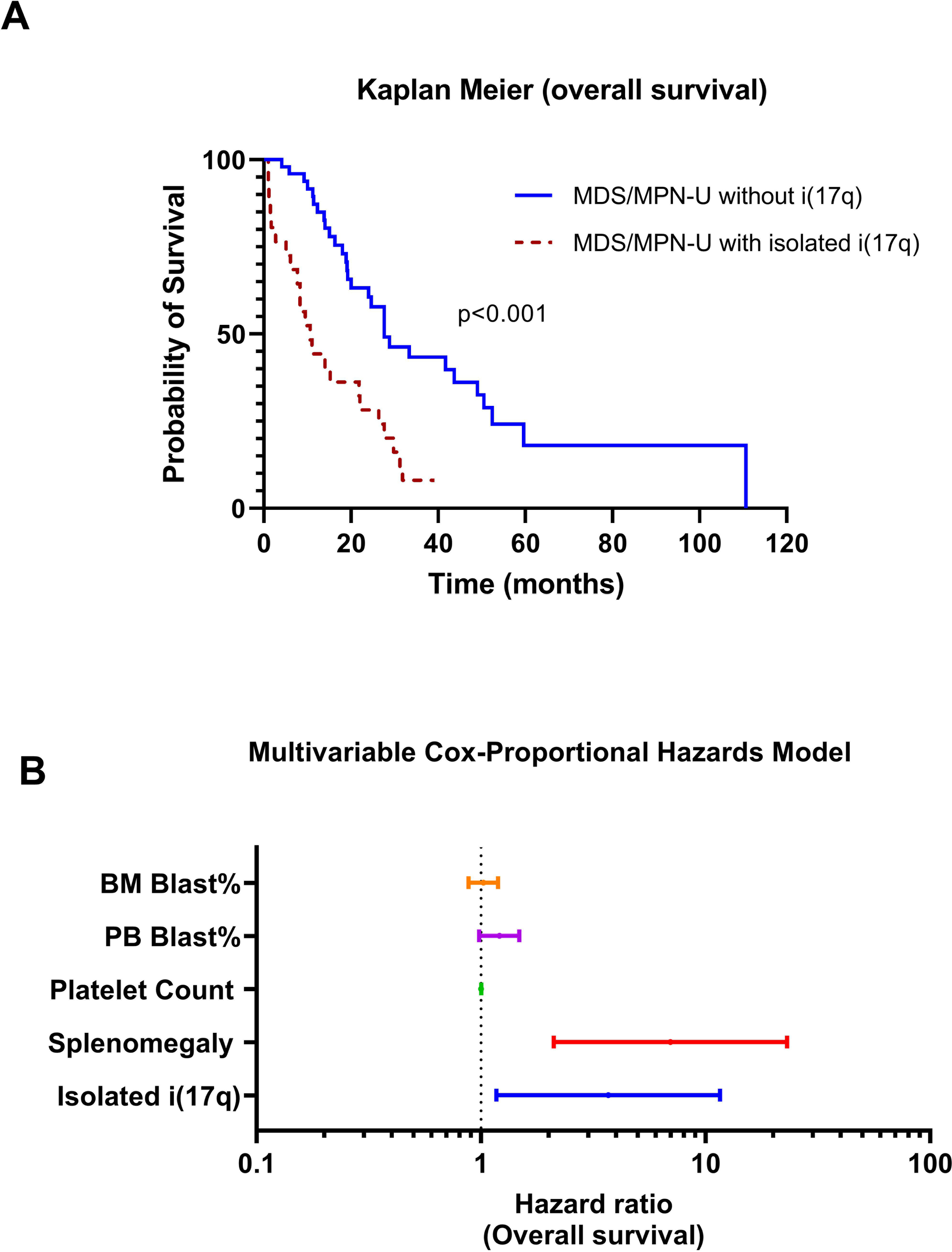
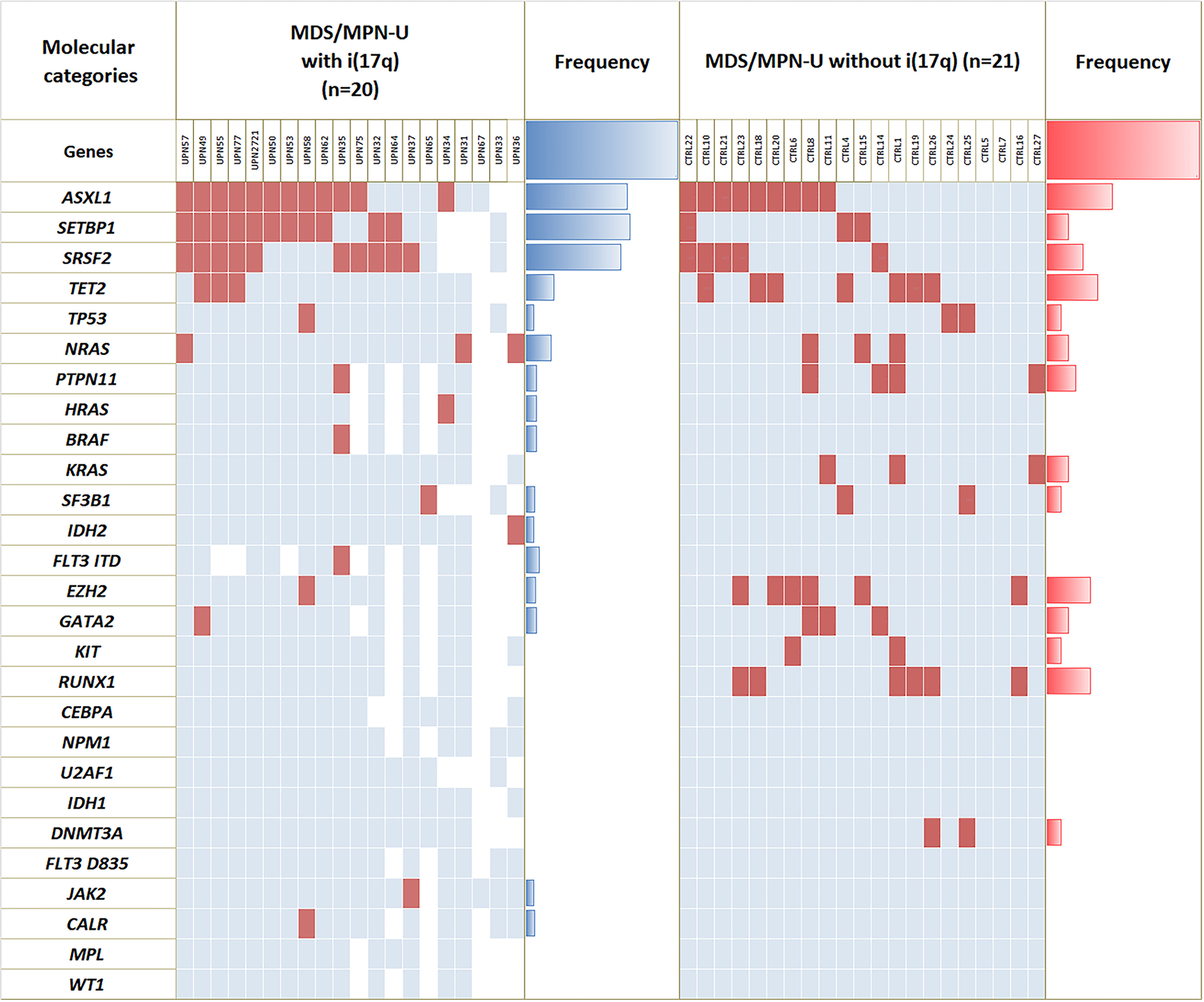
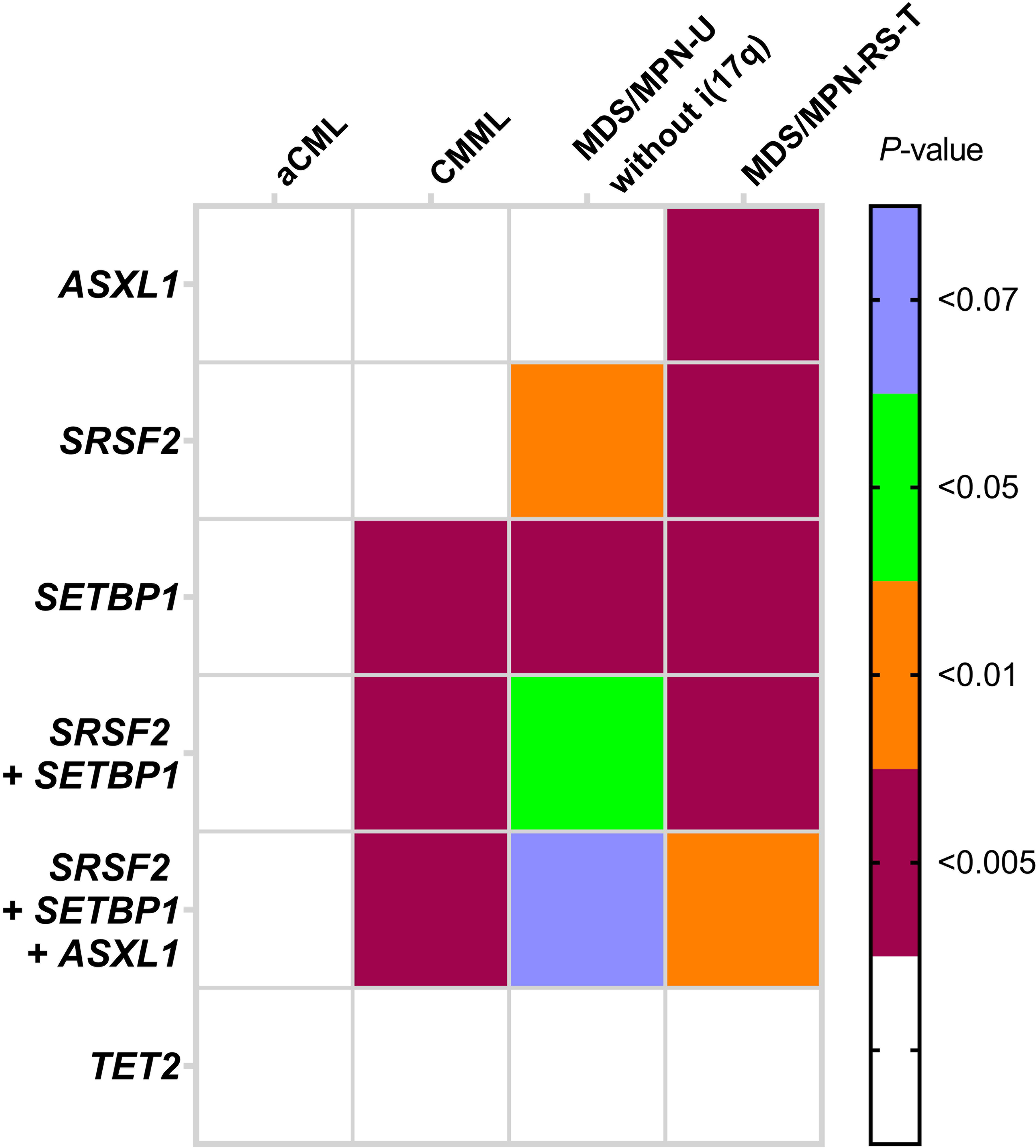
Classification of myeloid neoplasms with isolated isochromosome i(17q) [17p deletion with inherent monoallelic TP53 loss plus 17q duplication] is controversial. Most cases fall within the WHO unclassifiable myelodysplastic/myeloproliferative neoplasms (MDS/MPN-U) category. The uniformly dismal outcomes warrant better understanding of this entity. We undertook a multi-institutional retrospective study of 92 adult MDS/MPN-U cases from eight institutions. Twenty-nine (32%) patients had isolated i(17q) [MDS/MPN-i(17q)]. Compared to MDS/MPN without i(17q), MDS/MPN-i(17q) patients were significantly younger, had lower platelet and absolute neutrophil counts, and higher frequency of splenomegaly and circulating blasts. MDS/MPN-i(17q) cases showed frequent bilobed neutrophils (75% vs. 23%; P = 0.03), hypolobated megakaryocytes (62% vs. 20%; P = 0.06), and a higher frequency of SETBP1 (69% vs. 5%; P = 0.002) and SRSF2 (63% vs. 5%; P = 0.006) mutations that were frequently co-existent (44% vs. 0%; P = 0.01). TP53 mutations were rare. The mutation profile of MDS/MPN-U-i(17q) was similar to other myeloid neoplasms with i(17q) including atypical chronic myeloid leukemia, chronic myelomonocytic leukemia, myelodysplastic/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis, myelodysplastic syndrome and acute myeloid leukemia, with frequent concomitant SETBP1/SRSF2 mutations observed across all the diagnostic entities. Over a median follow-up of 52 months, patients with MDS/MPN-i(17q) showed a shorter median overall survival (11 vs. 28 months; P < 0.001). The presence of i(17q) retained independent poor prognostic value in multivariable Cox-regression analysis [HR 3.686 (1.17-11.6); P = 0.026] along with splenomegaly. We suggest that MDS/MPN-i(17q) warrants recognition as a distinct subtype within the MDS/MPN-U category based on its unique clinico-biologic features and uniformly poor prognosis.
孤立性 i(17q) [17p 缺失伴固有单等位基因 TP53 缺失加 17q 重复] 的髓系肿瘤分类存在争议。大多数病例属于世界卫生组织未分类的骨髓增生异常/骨髓增殖性肿瘤 (MDS/MPN-U) 类别。一致的预后不良需要更好地了解这种实体。我们对来自八个机构的 92 例成人 MDS/MPN-U 病例进行了多机构回顾性研究。29 例(32%)患者存在孤立性 i(17q) [MDS/MPN-i(17q)]。与无 i(17q) 的 MDS/MPN 相比,MDS/MPN-i(17q) 患者明显更年轻,血小板和绝对中性粒细胞计数更低,脾肿大和循环中原始细胞的频率更高。MDS/MPN-i(17q) 病例表现为频繁的双核中性粒细胞(75%比 23%;P=0.03)、低叶巨核细胞(62%比 20%;P=0.06)和更高频率的 SETBP1(69%比 5%;P=0.002)和 SRSF2(63%比 5%;P=0.006)突变,这些突变经常同时存在(44%比 0%;P=0.01)。TP53 突变很少见。MDS/MPN-U-i(17q) 的突变谱与其他具有 i(17q) 的髓系肿瘤相似,包括不典型慢性髓性白血病、慢性髓单核细胞白血病、伴有环形铁幼粒细胞和血小板增多的骨髓增生异常/骨髓增殖性肿瘤、骨髓增生异常综合征和急性髓细胞白血病,在所有诊断实体中都观察到频繁伴随的 SETBP1/SRSF2 突变。在中位随访 52 个月后,MDS/MPN-i(17q) 患者的中位总生存期较短(11 个月比 28 个月;P<0.001)。在多变量 Cox 回归分析中,i(17q) 的存在保留了独立的不良预后价值[风险比 3.686(1.17-11.6);P=0.026],同时伴有脾肿大。我们建议,根据其独特的临床生物学特征和一致的不良预后,将 MDS/MPN-i(17q) 确认为 MDS/MPN-U 类别中的一个独特亚型。