Wei Chunling, Li Yan, Feng Xiaoxiao, Hu Zhulin, Paquet-Durand François, Jiao Kangwei
Kunming Medical University, Kunming, China.
Department of Ophthalmology, Affiliated Hospital of Yunnan University, Yunnan University, Kunming, China.
Front Genet. 2021 Oct 29;12:728791. doi: 10.3389/fgene.2021.728791. eCollection 2021.
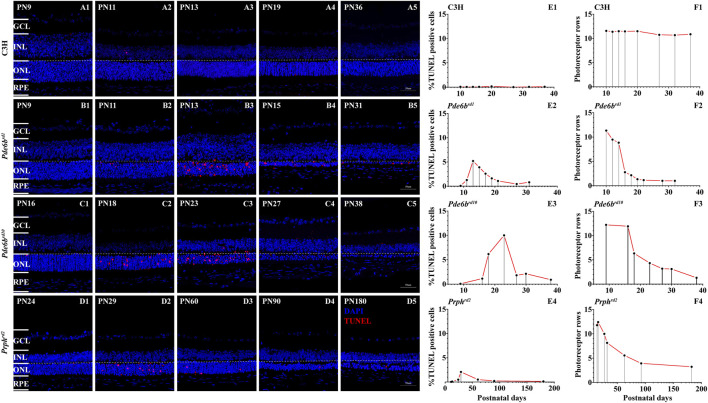
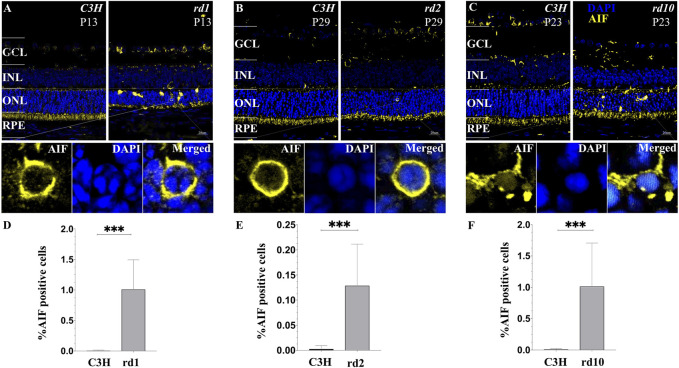
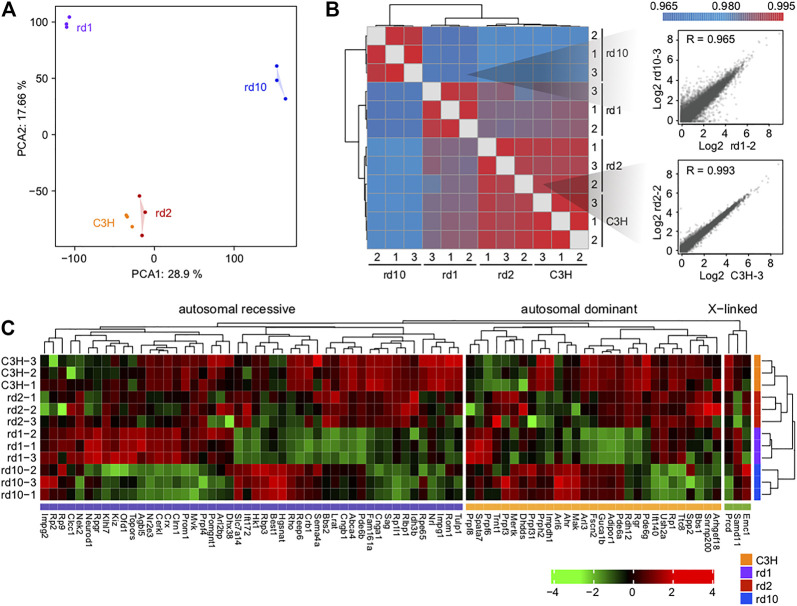
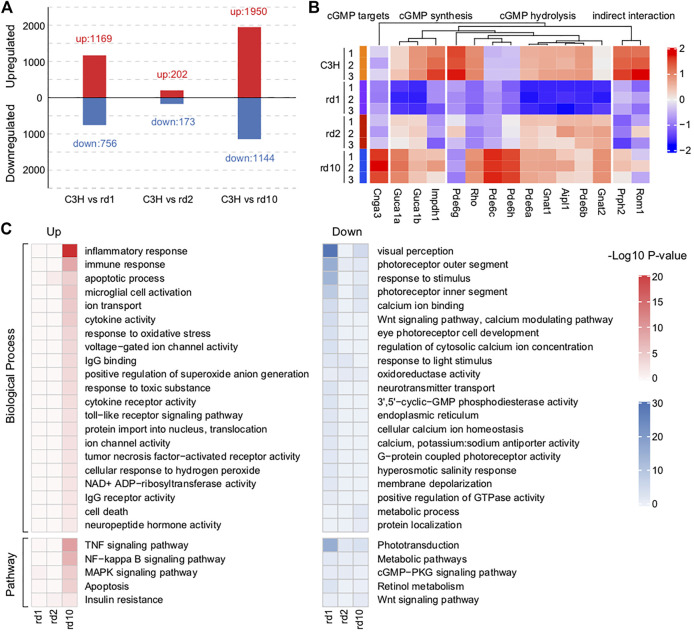
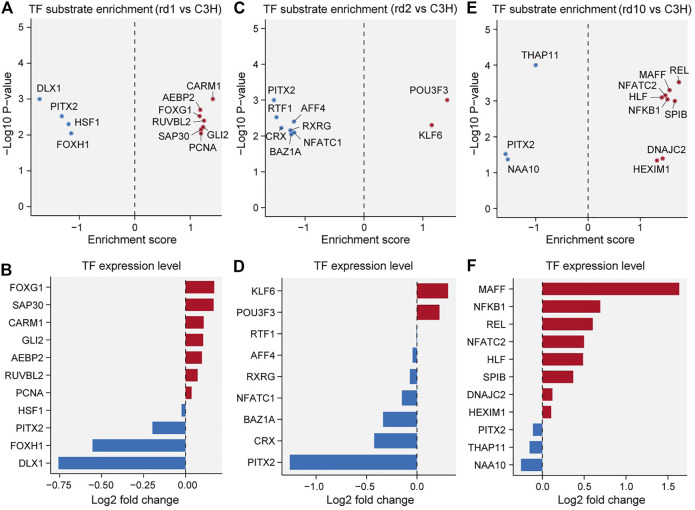
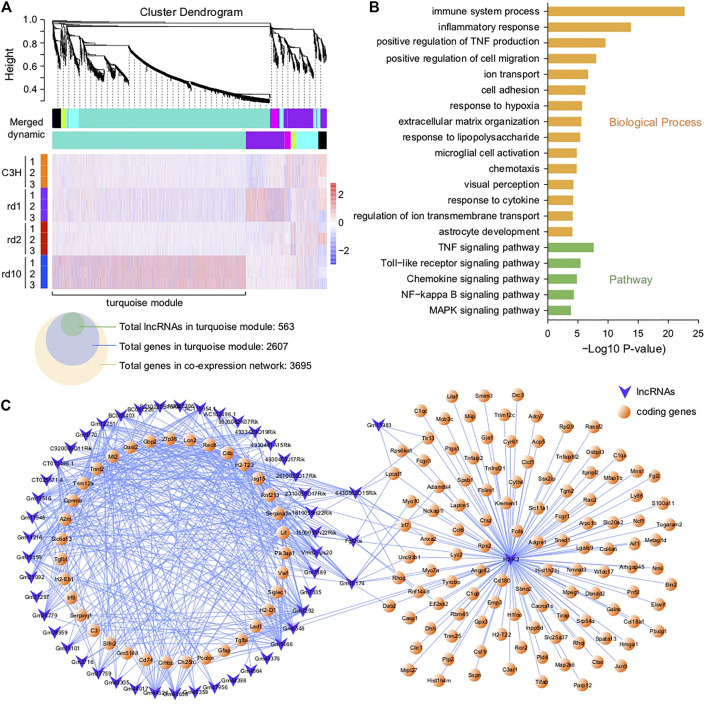
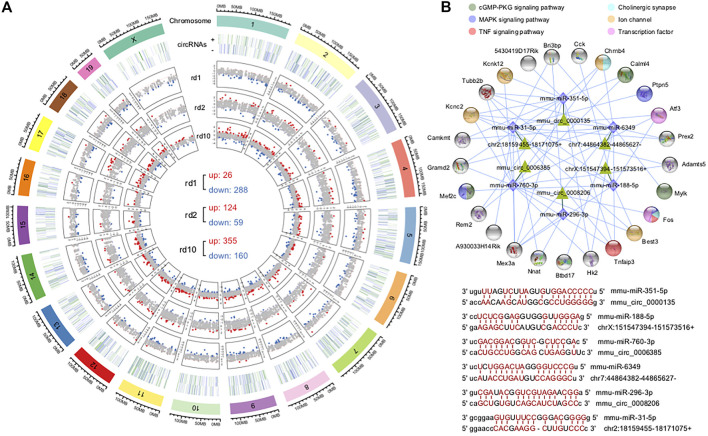
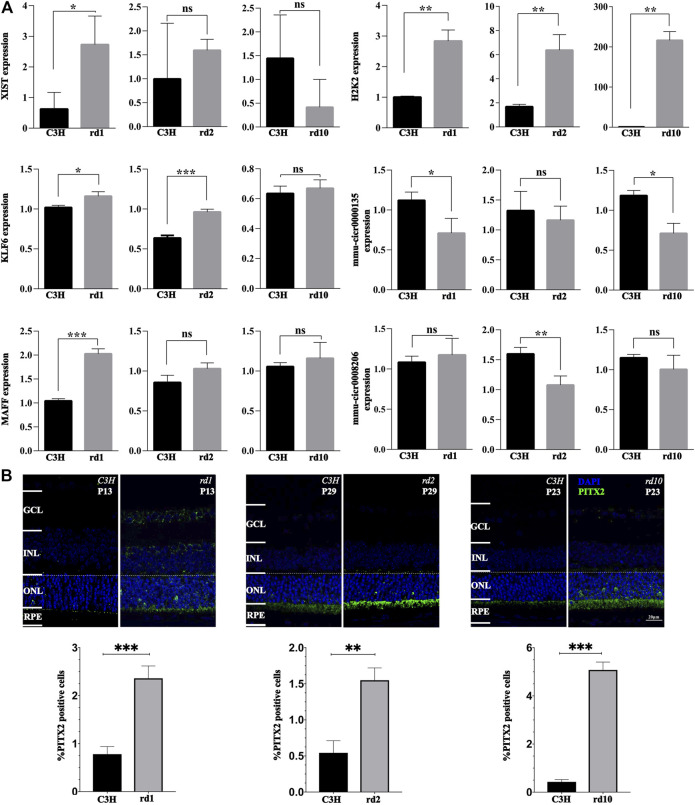
The present work investigated changes in the gene expression, molecular mechanisms, and pathogenesis of inherited retinal degeneration (RD) in three different disease models, to identify predictive biomarkers for their varied phenotypes and to provide a better scientific basis for their diagnosis, treatment, and prevention. Differentially expressed genes (DEGs) between retinal tissue from RD mouse models obtained during the photoreceptor cell death peak period ( at post-natal (PN) day 13, at PN23, at PN29) and retinal tissue from C3H wild-type mice were identified using Illumina high-throughput RNA-sequencing. Co-expression gene modules were identified using a combination of GO and KEGG enrichment analyses and gene co-expression network analysis. CircRNA-miRNA-mRNA network interactions were studied by genome-wide circRNA screening. mice had 1,926, 3,096, and 375 DEGs, respectively. Genes related to ion channels, stress, inflammatory processes, tumor necrosis factor (TNF) production, and microglial cell activation were up-regulated, while genes related to endoplasmic reticulum regulation, metabolism, and homeostasis were down-regulated. Differential expression of transcription factors and non-coding RNAs generally implicated in other human diseases was detected (e.g., glaucoma, diabetic retinopathy, and inherited retinal degeneration). CircRNA-miRNA-mRNA network analysis indicated that these factors may be involved in photoreceptor cell death. Moreover, excessive cGMP accumulation causes photoreceptor cell death, and cGMP-related genes were generally affected by different pathogenic gene mutations. We screened genes and pathways related to photoreceptor cell death. Additionally, up-stream regulatory factors, such as transcription factors and non-coding RNA and their interaction networks were analyzed. Furthermore, RNAs involved in RD were functionally annotated. Overall, this study lays a foundation for future studies on photoreceptor cell death mechanisms.
本研究在三种不同的疾病模型中,对遗传性视网膜变性(RD)的基因表达变化、分子机制及发病机制进行了研究,以确定其不同表型的预测生物标志物,并为其诊断、治疗及预防提供更好的科学依据。利用Illumina高通量RNA测序技术,鉴定了在光感受器细胞死亡高峰期(出生后(PN)第13天、PN23天、PN29天)获得的RD小鼠模型视网膜组织与C3H野生型小鼠视网膜组织之间的差异表达基因(DEG)。通过GO和KEGG富集分析以及基因共表达网络分析相结合的方法,鉴定了共表达基因模块。通过全基因组环状RNA筛选,研究了环状RNA-微小RNA-信使RNA网络相互作用。 小鼠分别有1926个、3096个和375个DEG。与离子通道、应激、炎症过程、肿瘤坏死因子(TNF)产生及小胶质细胞激活相关的基因上调,而与内质网调节、代谢及稳态相关的基因下调。检测到通常与其他人类疾病相关的转录因子和非编码RNA的差异表达(如青光眼、糖尿病性视网膜病变及遗传性视网膜变性)。环状RNA-微小RNA-信使RNA网络分析表明,这些因素可能参与光感受器细胞死亡。此外,过量的环磷酸鸟苷(cGMP)积累导致光感受器细胞死亡,且与cGMP相关的基因通常受不同致病基因突变的影响。我们筛选了与光感受器细胞死亡相关的基因和通路。此外,还分析了上游调节因子,如转录因子和非编码RNA及其相互作用网络。此外,对参与RD的RNA进行了功能注释。总体而言,本研究为未来关于光感受器细胞死亡机制的研究奠定了基础。