Merico Daniele, Pasternak Yehonatan, Zarrei Mehdi, Higginbotham Edward J, Thiruvahindrapuram Bhooma, Scott Ori, Willett-Pachul Jessica, Grunebaum Eyal, Upton Julia, Atkinson Adelle, Kim Vy H D, Aliyev Elbay, Fakhro Khalid, Scherer Stephen W, Roifman Chaim M
The Centre for Applied Genomics (TCAG), Program in Genetics and Genome Biology, The Hospital for Sick Children, Toronto, M5G 0A4, ON, Canada.
Deep Genomics Inc., Toronto, M5G 1M1, ON, Canada.
NPJ Genom Med. 2021 Nov 18;6(1):96. doi: 10.1038/s41525-021-00263-z.
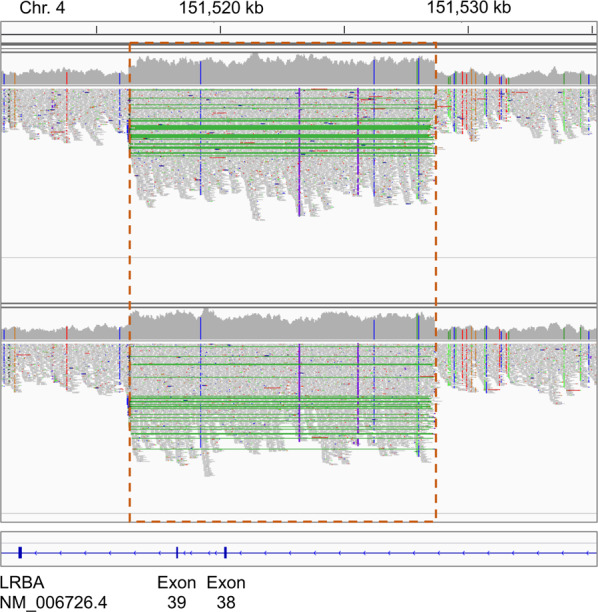
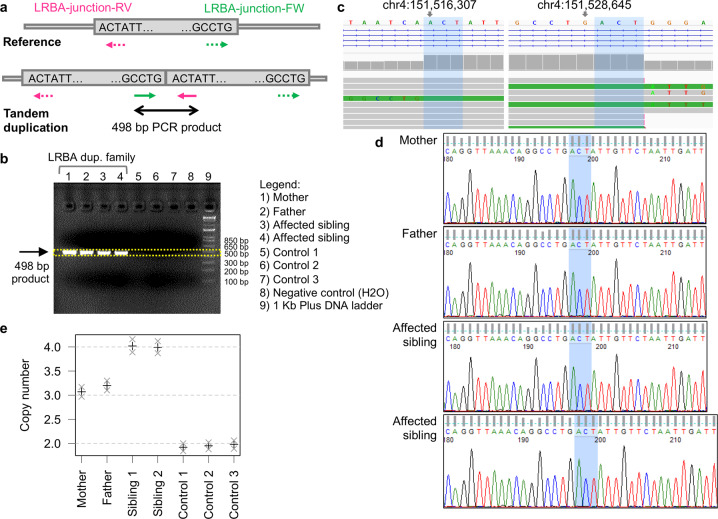
In more than one-third of primary immunodeficiency (PID) patients, extensive genetic analysis including whole-exome sequencing (WES) fails to identify the genetic defect. Whole-genome sequencing (WGS) is able to detect variants missed by other genomics platforms, enabling the molecular diagnosis of otherwise unresolved cases. Here, we report two siblings, offspring of consanguineous parents, who experienced similar severe events encompassing early onset of colitis, lymphoproliferation, and hypogammaglobulinemia, typical of lipopolysaccharide-responsive and beige-like anchor (LRBA) or cytotoxic T lymphocyte antigen 4 (CTLA4) deficiencies. Gene-panel sequencing, comparative genomic hybridization (CGH) array, and WES failed to reveal a genetic aberration in relevant genes. WGS of these patients detected a 12.3 kb homozygous tandem duplication that was absent in control cohorts and is predicted to disrupt the reading frame of the LRBA gene. The variant was validated by PCR and Sanger sequencing, demonstrating the presence of the junction between the reference and the tandem-duplicated sequence. Droplet digital PCR (ddPCR) further confirmed the copy number in the unaffected parents (CN = 3, heterozygous) and affected siblings (CN = 4, homozygous), confirming the expected segregation pattern. In cases of suspected inherited immunodeficiency, WGS may reveal a mutation when other methods such as microarray and WES analysis failed to detect an aberration.
在超过三分之一的原发性免疫缺陷(PID)患者中,包括全外显子组测序(WES)在内的广泛基因分析未能识别出基因缺陷。全基因组测序(WGS)能够检测出其他基因组学平台遗漏的变异,从而对原本无法确诊的病例进行分子诊断。在此,我们报告了一对近亲父母的子女,这两名兄弟姐妹经历了相似的严重事件,包括早期发作的结肠炎、淋巴细胞增殖和低丙种球蛋白血症,这些都是脂多糖反应性和米色样锚定蛋白(LRBA)或细胞毒性T淋巴细胞抗原4(CTLA4)缺陷的典型症状。基因panel测序、比较基因组杂交(CGH)阵列和WES均未发现相关基因存在遗传畸变。对这些患者进行WGS检测到一个12.3 kb的纯合串联重复序列,该序列在对照队列中不存在,预计会破坏LRBA基因的阅读框。通过PCR和桑格测序对该变异进行了验证,证实了参考序列与串联重复序列之间连接点的存在。液滴数字PCR(ddPCR)进一步确认了未受影响父母(拷贝数=3,杂合子)和受影响兄弟姐妹(拷贝数=4,纯合子)中的拷贝数,证实了预期的遗传分离模式。在疑似遗传性免疫缺陷的病例中,当微阵列和WES分析等其他方法未能检测到畸变时,WGS可能会揭示出突变。