Song Yunjia, Song Jiaru, Zhu Zhigang, Peng Hanlin, Ding Xiang, Yang Fuquan, Li Kun, Yu Xiaoqi, Yang Guosheng, Tao Yinghong, Bu Dingfang, Tang Chaoshu, Huang Yaqian, Du Junbao, Jin Hongfang
Department of Pediatrics, Peking University First Hospital, Beijing, China.
Key Laboratory of Protein and Peptide Pharmaceuticals & Laboratory of Proteomics, Institute of Biophysics, Chinese Academy of Sciences, Beijing, 100101, China.
Redox Biol. 2021 Nov 18;48:102192. doi: 10.1016/j.redox.2021.102192.
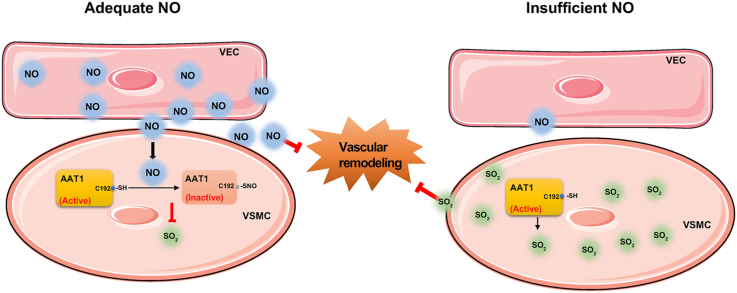
This study aimed to determine the communicational pattern of gaseous signaling molecules sulfur dioxide (SO) and nitric oxide (NO) between vascular endothelial cells (VECs) and vascular smooth muscle cells (VSMCs), and elucidate the compensatory role and significance of endogenous SO in the development of hypertension due to NO deficiency.
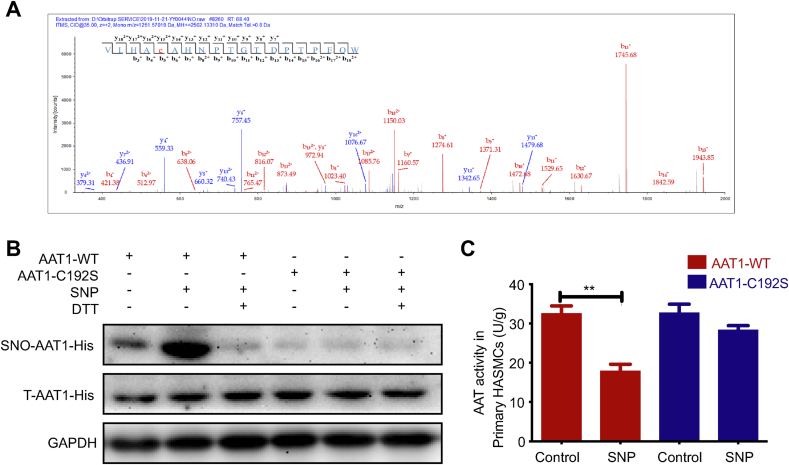
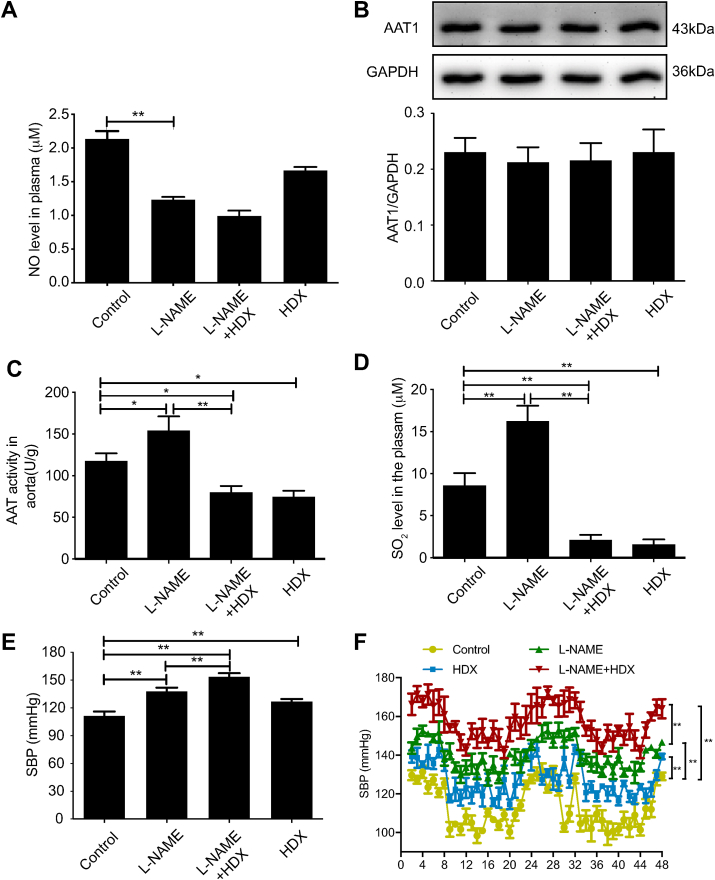
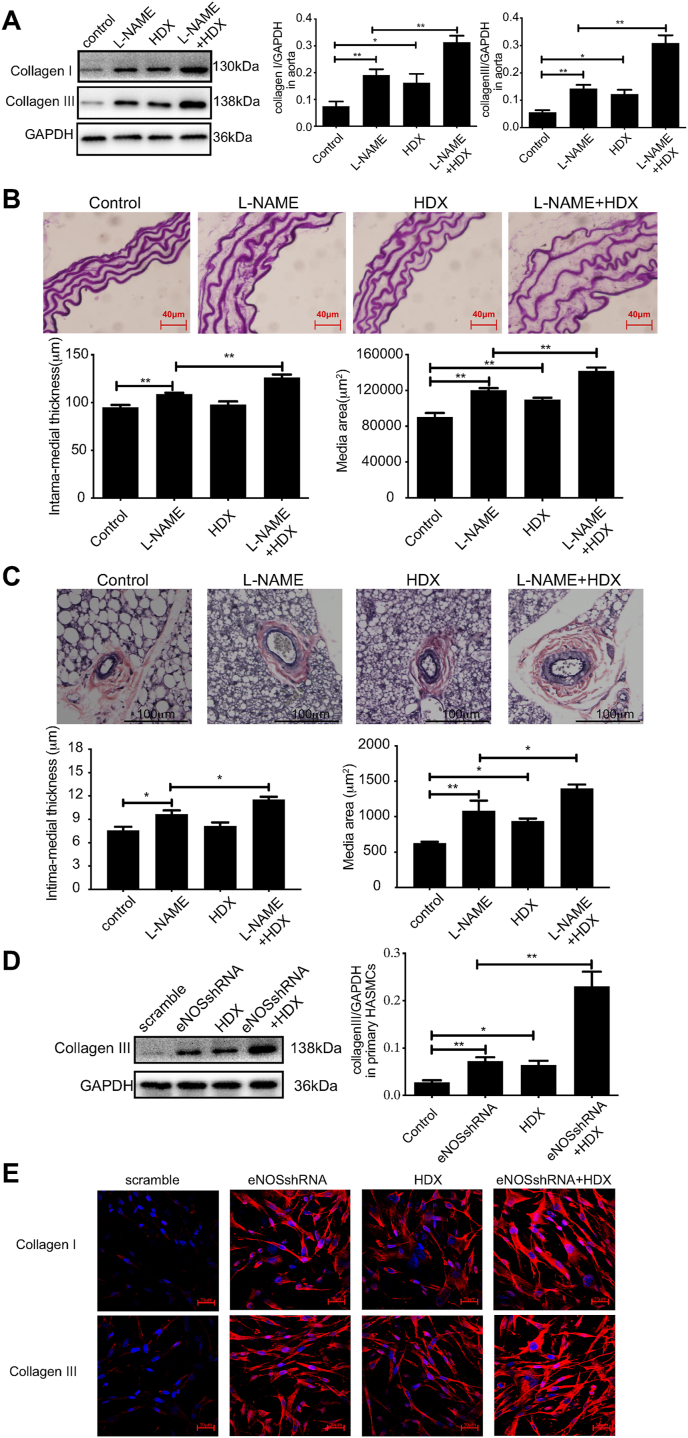
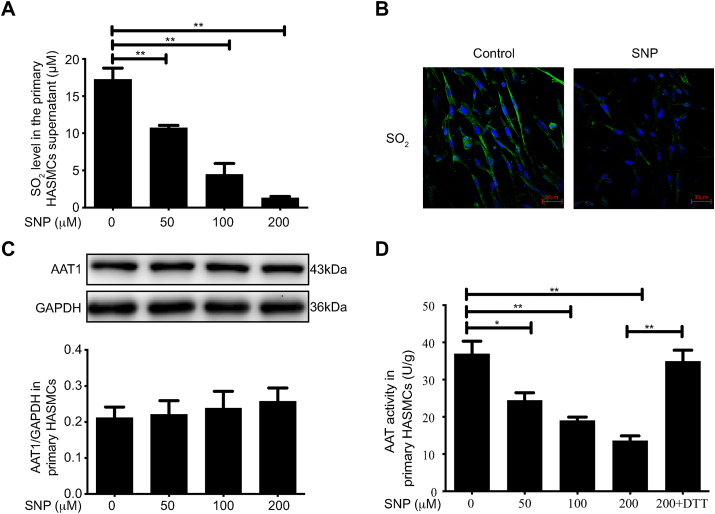
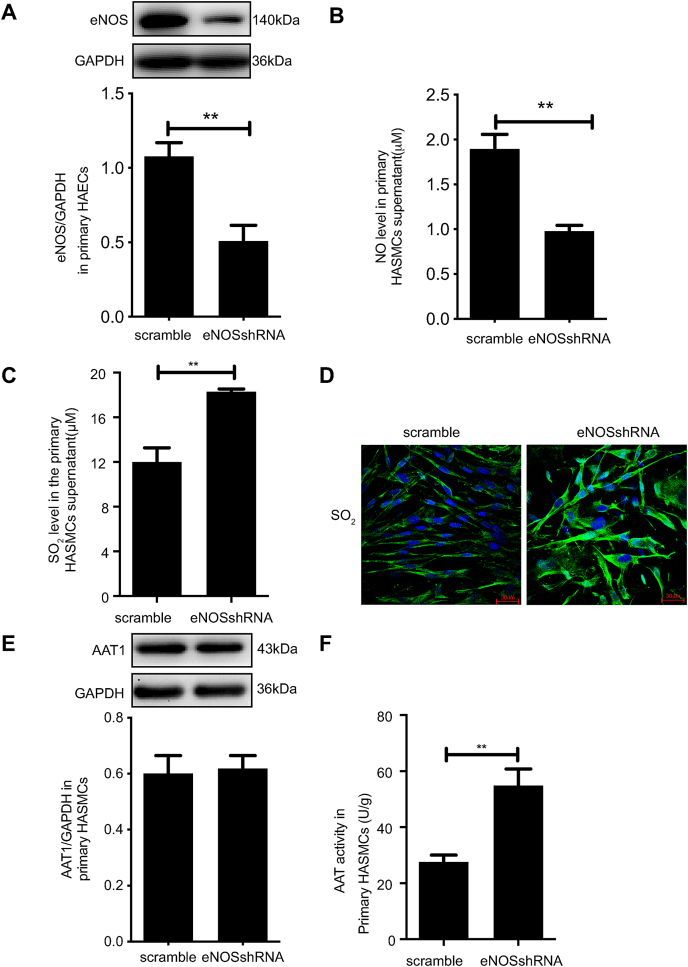
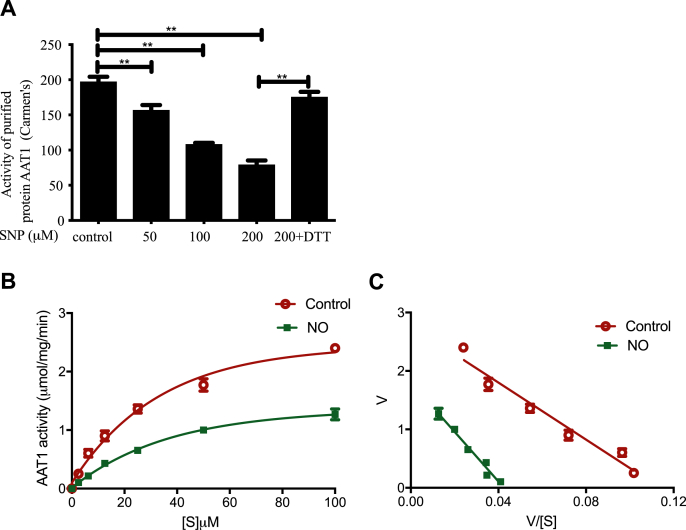
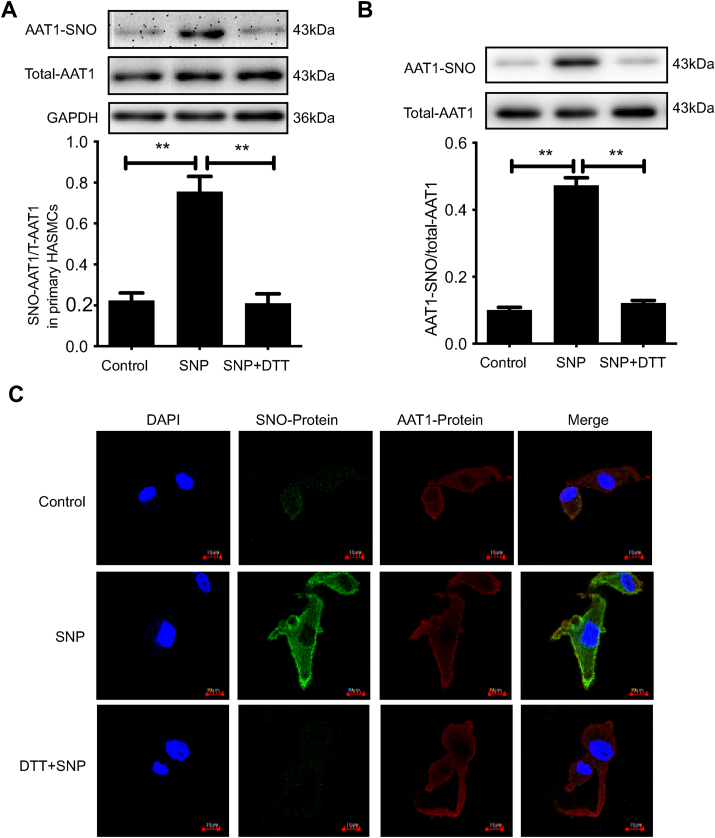
Blood pressure was monitored by the tail-cuff and implantable physiological signal telemetry in L-nitro-arginine methyl ester (l-NAME)-induced hypertensive mice, and structural alterations of mouse aortic vessels were detected by the elastic fiber staining method. l-NAME-treated mice showed decreased plasma NO levels, increased SO levels, vascular remodeling, and increased blood pressure, and application of l-aspartate-β-hydroxamate, which inhibits SO production, further aggravated vascular structural remodeling and increased blood pressure. Moreover, in a co-culture system of HAECs and HASMCs, NO from HAECs did not influence aspartate aminotransferase (AAT)1 protein expression but decreased AAT1 activity in HASMCs, thereby resulting in the inhibition of endogenous SO production. Furthermore, NO promoted S-nitrosylation of AAT1 protein in HASMCs and purified AAT1 protein. Liquid chromatography with tandem mass spectrometry showed that the Cys192 site of AAT1 purified protein was modified by S-nitrosylation. In contrast, dithiothreitol or C192S mutations in HASMCs blocked NO-induced AAT1 S-nitrosylation and restored AAT1 enzyme activity.
Endothelium-derived NO inhibits AAT activity by nitrosylating AAT1 at the Cys192 site and reduces SO production in HASMCs. Our findings suggest that SO acts as a compensatory defense system to antagonize vascular structural remodeling and hypertension when the endogenous NO pathway is disturbed.
本研究旨在确定血管内皮细胞(VECs)与血管平滑肌细胞(VSMCs)之间气态信号分子二氧化硫(SO)和一氧化氮(NO)的通信模式,并阐明内源性SO在因NO缺乏导致的高血压发展中的代偿作用及意义。
通过尾套法和植入式生理信号遥测技术监测L-硝基精氨酸甲酯(L-NAME)诱导的高血压小鼠的血压,并采用弹性纤维染色法检测小鼠主动脉血管的结构改变。L-NAME处理的小鼠血浆NO水平降低、SO水平升高、血管重塑且血压升高,应用抑制SO生成的L-天冬氨酸-β-羟肟酸进一步加重了血管结构重塑并升高了血压。此外,在人主动脉内皮细胞(HAECs)与人主动脉平滑肌细胞(HASMCs)的共培养体系中,HAECs产生的NO不影响HASMCs中天冬氨酸转氨酶(AAT)1蛋白表达,但降低了其活性,从而抑制了内源性SO的生成。此外,NO促进了HASMCs中AAT1蛋白和纯化的AAT1蛋白的S-亚硝基化。液相色谱-串联质谱分析表明,纯化的AAT1蛋白的Cys192位点发生了S-亚硝基化修饰。相反,HASMCs中的二硫苏糖醇或C192S突变可阻断NO诱导的AAT1 S-亚硝基化并恢复AAT1酶活性。
内皮源性NO通过在Cys192位点使AAT1亚硝基化来抑制AAT活性,并减少HASMCs中SO的生成。我们的研究结果表明,当内源性NO途径受到干扰时,SO作为一种代偿性防御系统可拮抗血管结构重塑和高血压。