Department of Medicine, Division of Endocrinology, David Geffen School of Medicine at UCLA, 650 Charles E. Young Dr., Los Angeles, CA 90095, USA; Department of Molecular Medicine, Faculty of Medicine, Universite Laval, Quebec City G1V 0A6, Canada.
Department of Medicine, Division of Endocrinology, David Geffen School of Medicine at UCLA, 650 Charles E. Young Dr., Los Angeles, CA 90095, USA.
Mol Metab. 2022 Jan;55:101403. doi: 10.1016/j.molmet.2021.101403. Epub 2021 Nov 23.
The contribution of beta-cell dysfunction to type 2 diabetes (T2D) is not restricted to insulinopenia in the late stages of the disease. Elevated fasting insulinemia in normoglycemic humans is a major factor predicting the onset of insulin resistance and T2D, demonstrating an early alteration of beta-cell function in T2D. Moreover, an early and chronic increase in fasting insulinemia contributes to insulin resistance in high-fat diet (HFD)-fed mice. However, whether there are genetic factors that promote beta-cell-initiated insulin resistance remains undefined. Human variants of the mitochondrial transporter ABCB10, which regulates redox by increasing bilirubin synthesis, have been associated with an elevated risk of T2D. The effects of T2D ABCB10 variants on ABCB10 expression and the actions of ABCB10 in beta-cells are unknown.
The expression of beta-cell ABCB10 was analyzed in published transcriptome datasets from human beta-cells carrying the T2D-risk ABCB10 variant. Insulin sensitivity, beta-cell proliferation, and secretory function were measured in beta-cell-specific ABCB10 KO mice (Ins1-Abcb10). The short-term role of beta-cell ABCB10 activity on glucose-stimulated insulin secretion (GSIS) was determined in isolated islets.
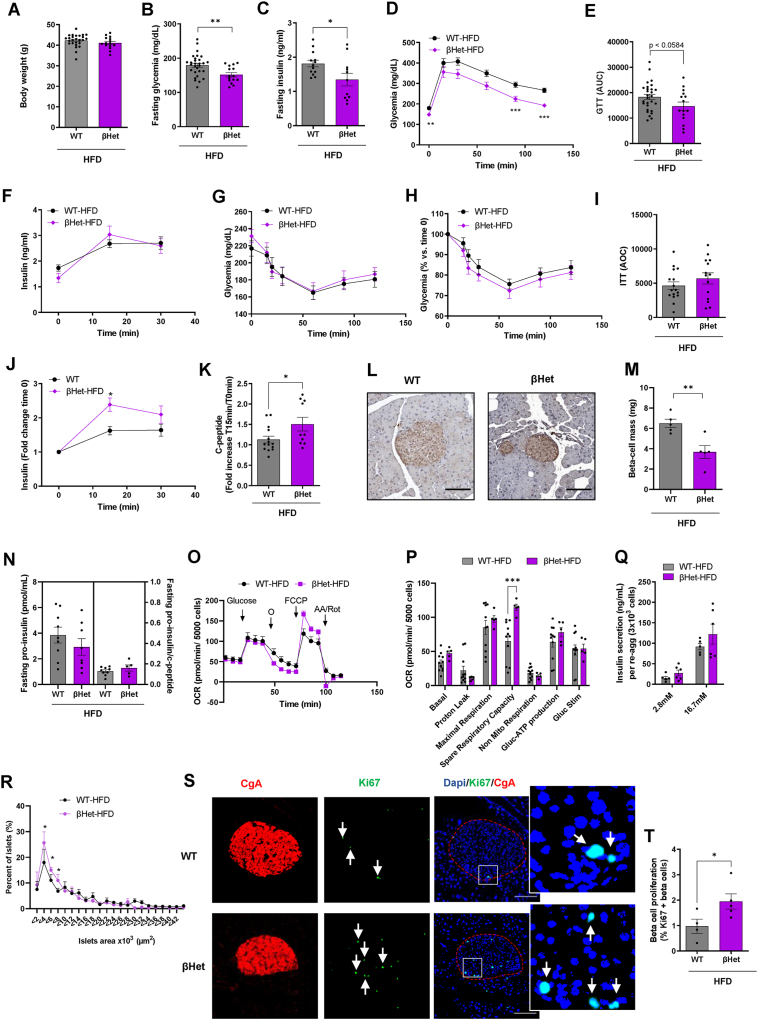
Carrying the T2Drisk allele G of ABCB10 rs348330 variant was associated with increased ABCB10 expression in human beta-cells. Constitutive deletion of Abcb10 in beta-cells protected mice from hyperinsulinemia and insulin resistance by limiting HFD-induced beta-cell expansion. An early limitation in GSIS and HO-mediated signaling caused by elevated ABCB10 activity can initiate an over-compensatory expansion of beta-cell mass in response to HFD. Accordingly, increasing ABCB10 expression was sufficient to limit GSIS capacity. In health, ABCB10 protein was decreased during islet maturation, with maturation restricting beta-cell proliferation and elevating GSIS. Finally, ex-vivo and short-term deletion of ABCB10 in islets isolated from HFD-fed mice increased HO and GSIS, which was reversed by bilirubin treatments.
Beta-cell ABCB10 is required for HFD to induce insulin resistance in mice by amplifying beta-cell mass expansion to maladaptive levels that cause fasting hyperinsulinemia.
β细胞功能障碍对 2 型糖尿病(T2D)的贡献不仅限于疾病晚期的胰岛素分泌不足。在血糖正常的人群中,空腹胰岛素升高是预测胰岛素抵抗和 T2D 发病的主要因素,表明 T2D 中β细胞功能的早期改变。此外,空腹胰岛素的早期和慢性升高可导致高脂肪饮食(HFD)喂养的小鼠胰岛素抵抗。然而,是否存在促进β细胞起始胰岛素抵抗的遗传因素尚不清楚。调节胆红素合成从而增加氧化还原的线粒体转运蛋白 ABCB10 的人类变体与 T2D 的风险增加有关。T2D ABCB10 变体对 ABCB10 表达的影响以及 ABCB10 在β细胞中的作用尚不清楚。
分析了携带 T2D 风险 ABCB10 变体的人β细胞中已发表的转录组数据集β细胞 ABCB10 的表达。在β细胞特异性 Abcb10 敲除(Ins1-Abcb10)小鼠中测量胰岛素敏感性、β细胞增殖和分泌功能。在分离的胰岛中确定β细胞 ABCB10 活性对葡萄糖刺激的胰岛素分泌(GSIS)的短期作用。
携带 ABCB10 rs348330 变体的 T2D 风险等位基因 G 与人类β细胞中 ABCB10 表达增加有关。β细胞中 Abcb10 的组成性缺失通过限制 HFD 诱导的β细胞扩张,使小鼠免受高胰岛素血症和胰岛素抵抗的影响。由于 ABCB10 活性升高导致的早期 GSIS 和 HO 介导的信号转导受限,可能会引发β细胞质量的过度代偿性扩张,以应对 HFD。因此,增加 ABCB10 表达足以限制 GSIS 能力。在健康状态下,胰岛成熟过程中 ABCB10 蛋白减少,成熟限制β细胞增殖并升高 GSIS。最后,从 HFD 喂养的小鼠分离的胰岛中进行离体和短期 Abcb10 缺失增加了 HO 和 GSIS,这可被胆红素处理逆转。
β细胞 ABCB10 是 HFD 在小鼠中诱导胰岛素抵抗所必需的,通过扩增β细胞质量扩张到适应不良的水平,导致空腹高胰岛素血症。