Department of Mathematical Sciences, School of Science, Zhejiang Sci-Tech University, Hangzhou, 310018, China.
BMC Genomics. 2021 Nov 29;22(1):860. doi: 10.1186/s12864-021-08101-3.
With single-cell RNA sequencing (scRNA-seq) methods, gene expression patterns at the single-cell resolution can be revealed. But as impacted by current technical defects, dropout events in scRNA-seq lead to missing data and noise in the gene-cell expression matrix and adversely affect downstream analyses. Accordingly, the true gene expression level should be recovered before the downstream analysis is carried out.
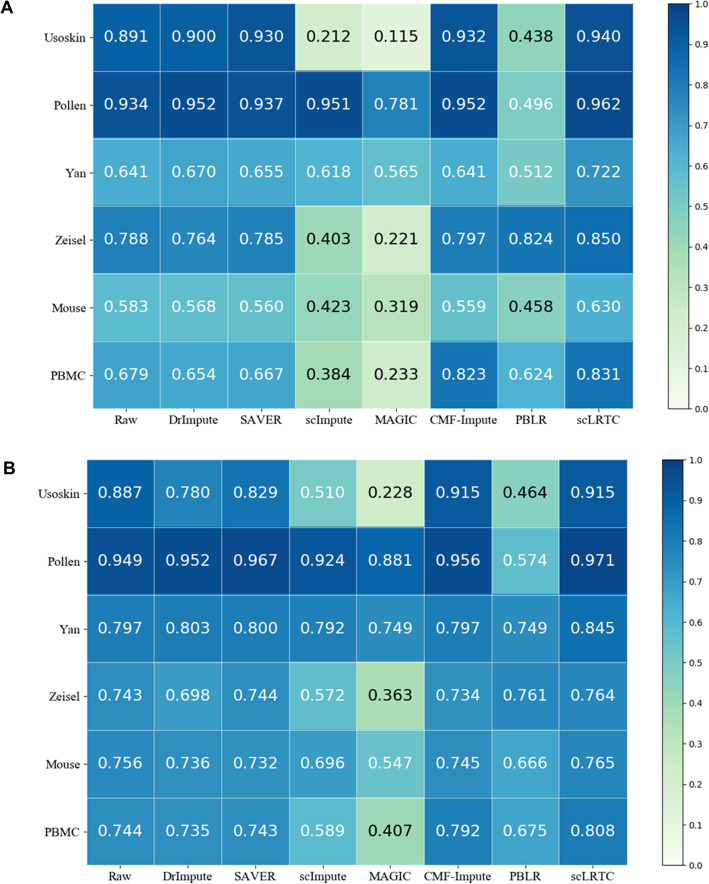
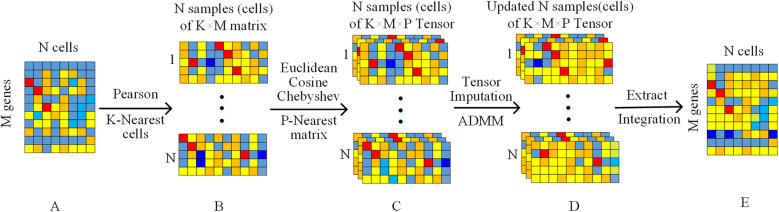
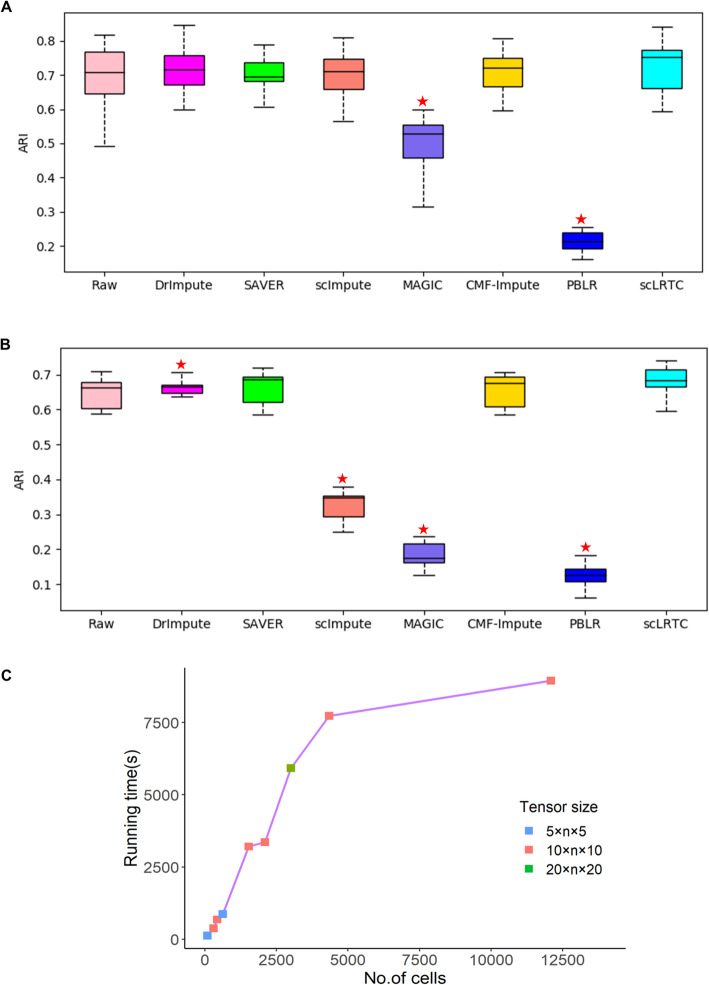
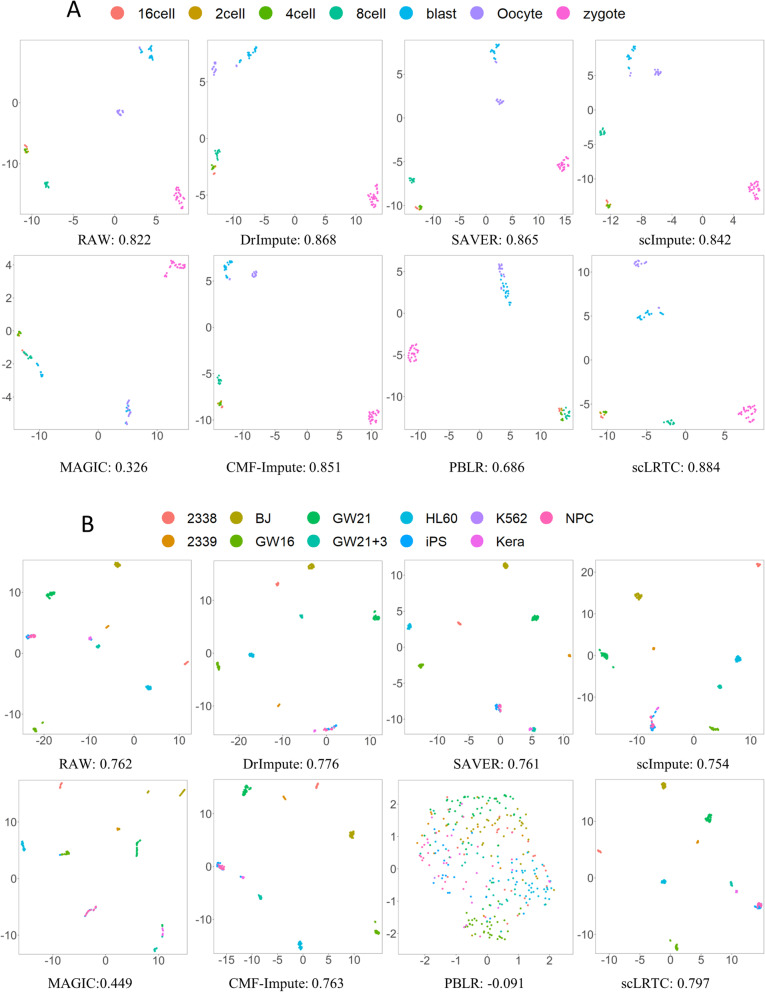
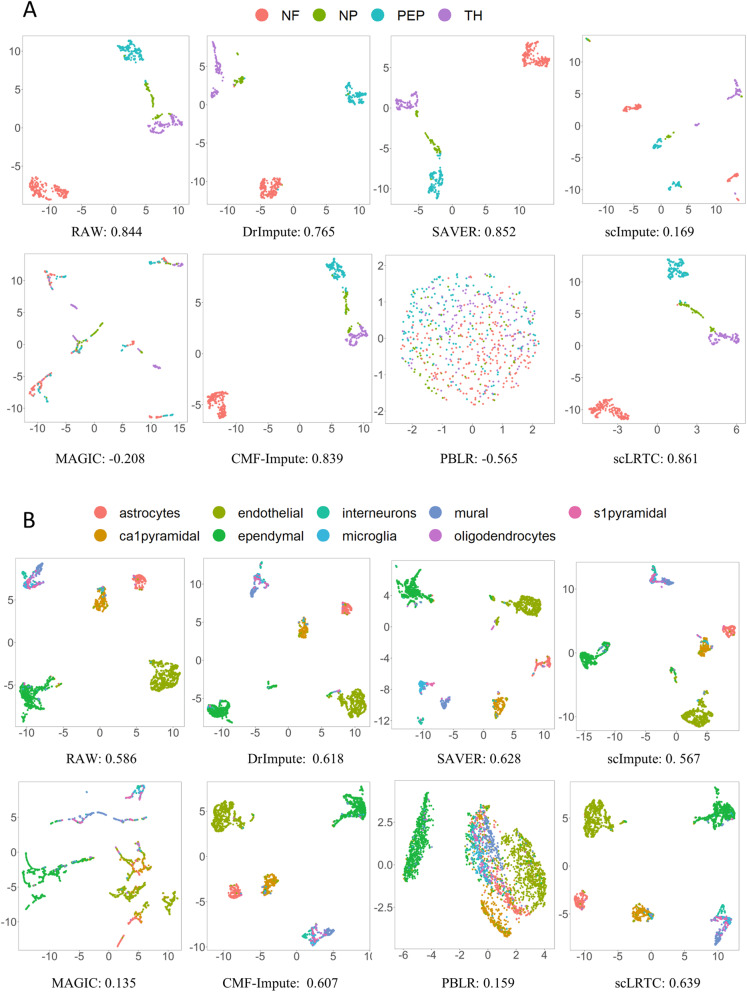
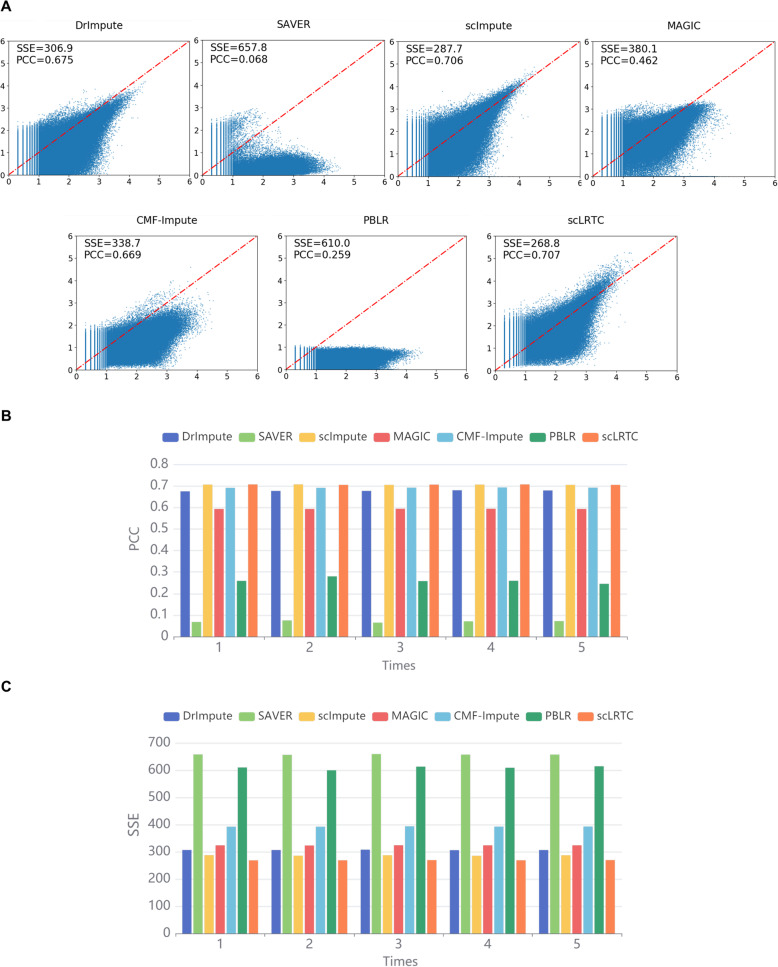
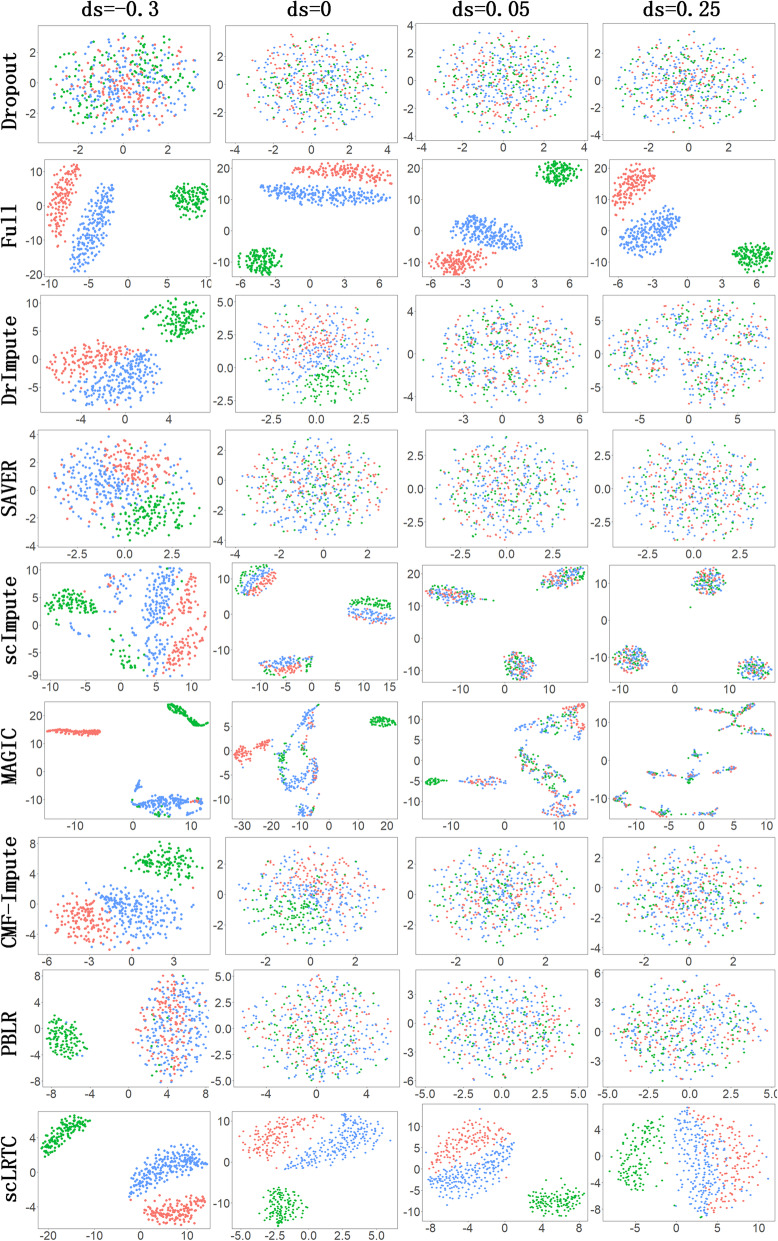
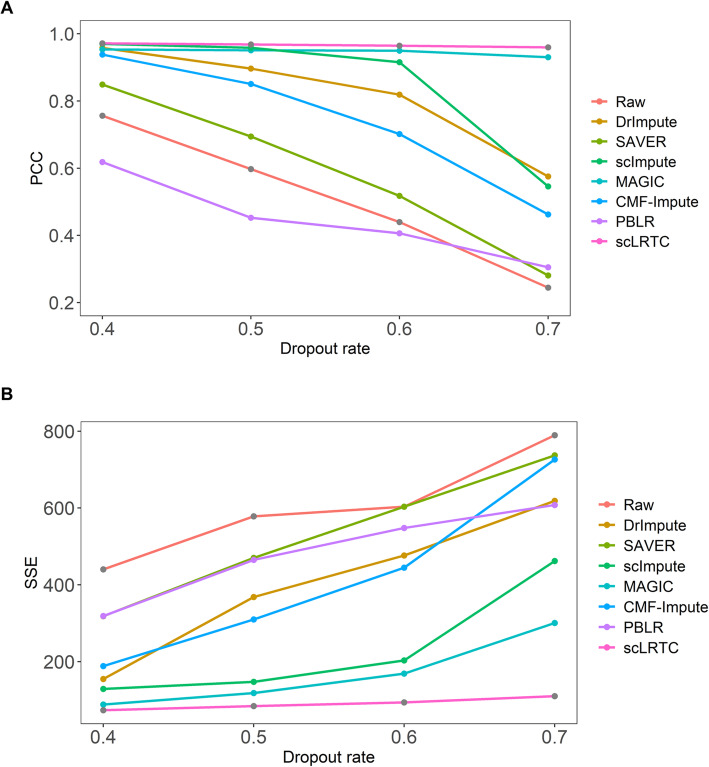
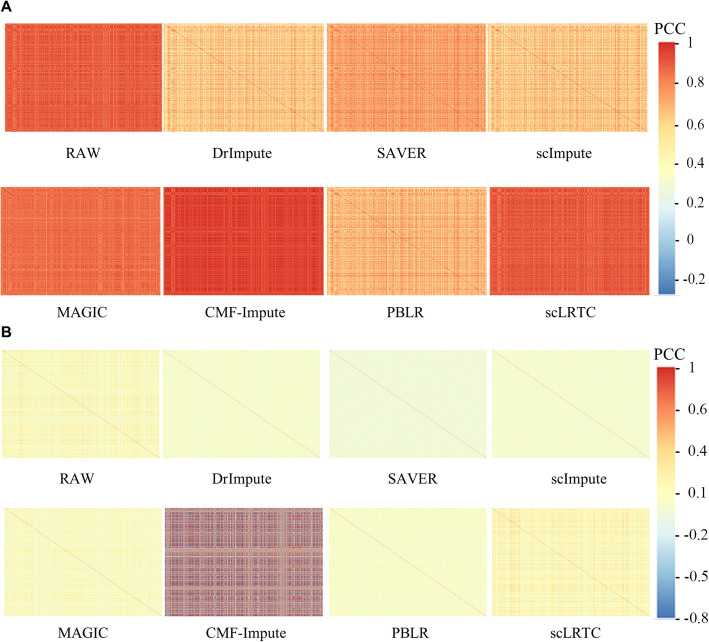
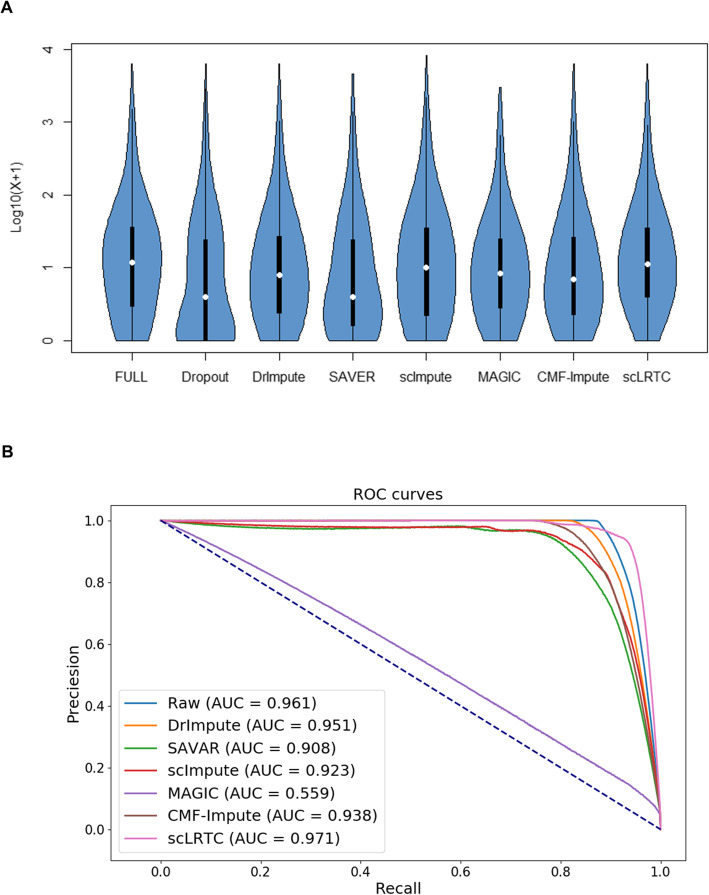
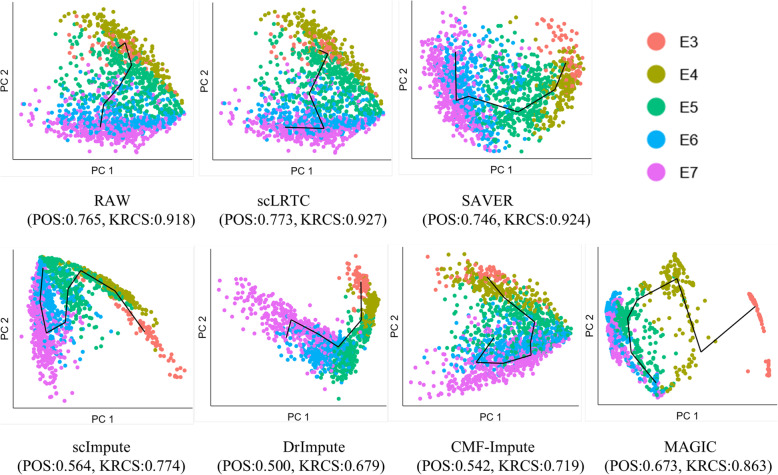
In this paper, a novel low-rank tensor completion-based method, termed as scLRTC, is proposed to impute the dropout entries of a given scRNA-seq expression. It initially exploits the similarity of single cells to build a third-order low-rank tensor and employs the tensor decomposition to denoise the data. Subsequently, it reconstructs the cell expression by adopting the low-rank tensor completion algorithm, which can restore the gene-to-gene and cell-to-cell correlations. ScLRTC is compared with other state-of-the-art methods on simulated datasets and real scRNA-seq datasets with different data sizes. Specific to simulated datasets, scLRTC outperforms other methods in imputing the dropouts closest to the original expression values, which is assessed by both the sum of squared error (SSE) and Pearson correlation coefficient (PCC). In terms of real datasets, scLRTC achieves the most accurate cell classification results in spite of the choice of different clustering methods (e.g., SC3 or t-SNE followed by K-means), which is evaluated by using adjusted rand index (ARI) and normalized mutual information (NMI). Lastly, scLRTC is demonstrated to be also effective in cell visualization and in inferring cell lineage trajectories.
a novel low-rank tensor completion-based method scLRTC gave imputation results better than the state-of-the-art tools. Source code of scLRTC can be accessed at https://github.com/jianghuaijie/scLRTC .
单细胞 RNA 测序 (scRNA-seq) 方法可以揭示单细胞分辨率下的基因表达模式。但由于当前技术缺陷的影响,scRNA-seq 中的缺失事件导致基因-细胞表达矩阵中出现缺失数据和噪声,从而对下游分析产生不利影响。因此,在进行下游分析之前,应该恢复真实的基因表达水平。
在本文中,提出了一种新的基于低秩张量补全的方法,称为 scLRTC,用于对给定的 scRNA-seq 表达中的缺失项进行插补。它首先利用单细胞之间的相似性构建三阶低秩张量,并采用张量分解对数据进行去噪。随后,通过采用低秩张量补全算法对细胞表达进行重构,从而恢复基因-基因和细胞-细胞之间的相关性。在模拟数据集和不同数据大小的真实 scRNA-seq 数据集上,与其他最先进的方法进行了比较。具体来说,在模拟数据集上,scLRTC 在插补最接近原始表达值的缺失值方面优于其他方法,这可以通过均方误差 (SSE) 和 Pearson 相关系数 (PCC) 来评估。就真实数据集而言,尽管选择了不同的聚类方法(例如,SC3 或 t-SNE 之后的 K-means),scLRTC 仍能获得最准确的细胞分类结果,这可以通过调整兰德指数 (ARI) 和归一化互信息 (NMI) 来评估。最后,证明了 scLRTC 也可以有效地进行细胞可视化和推断细胞谱系轨迹。
一种新的基于低秩张量补全的方法 scLRTC 给出的插补结果优于最先进的工具。scLRTC 的源代码可以在 https://github.com/jianghuaijie/scLRTC 上访问。