NCMIS, CEMS, RCSDS, Academy of Mathematics and Systems Science, Chinese Academy of Sciences, Beijing 100190, China.
School of Mathematical Sciences, University of Chinese Academy of Sciences, Beijing 100049, China.
J Mol Cell Biol. 2021 Apr 10;13(1):29-40. doi: 10.1093/jmcb/mjaa052.
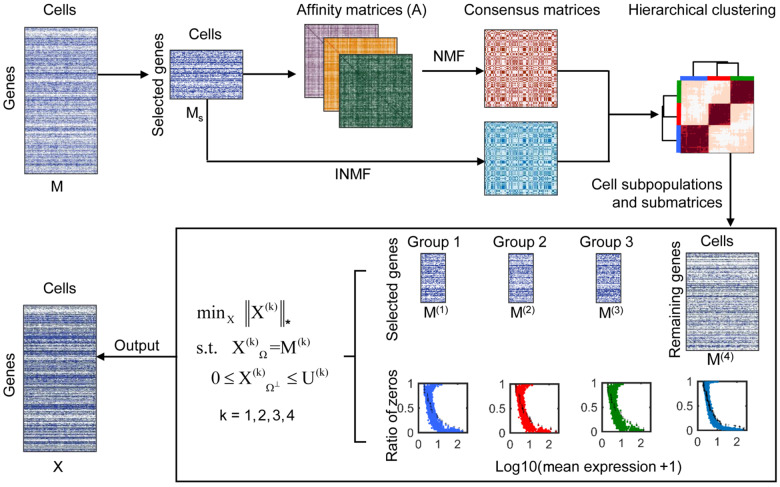
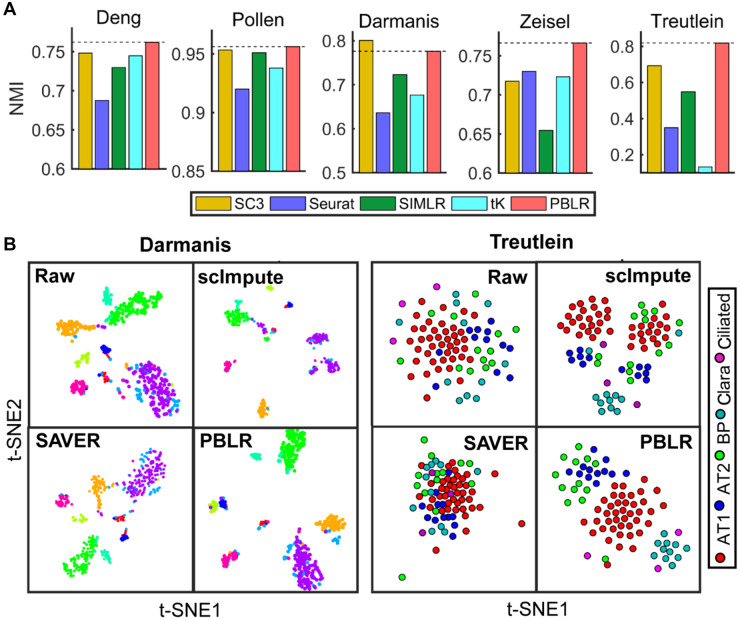
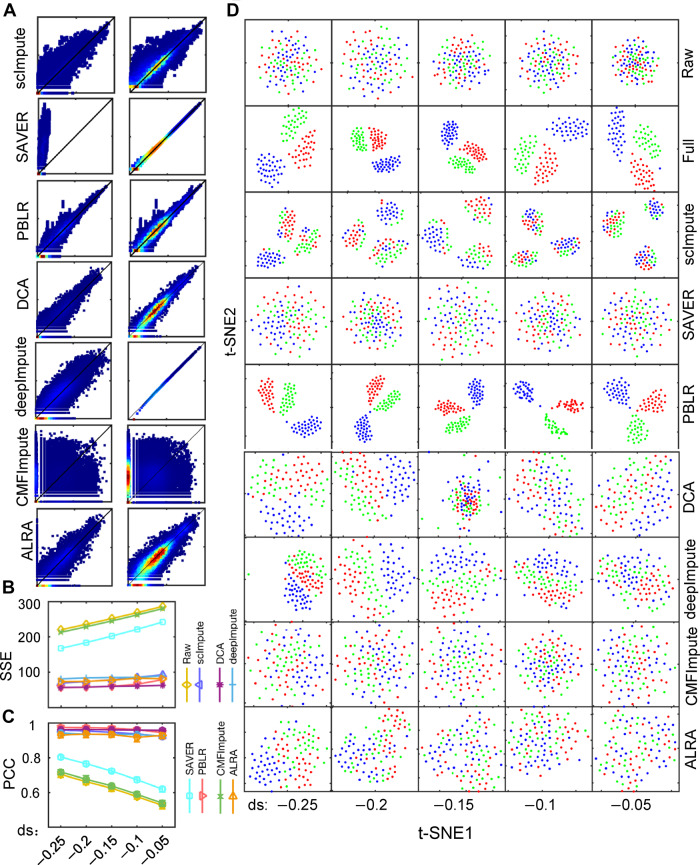
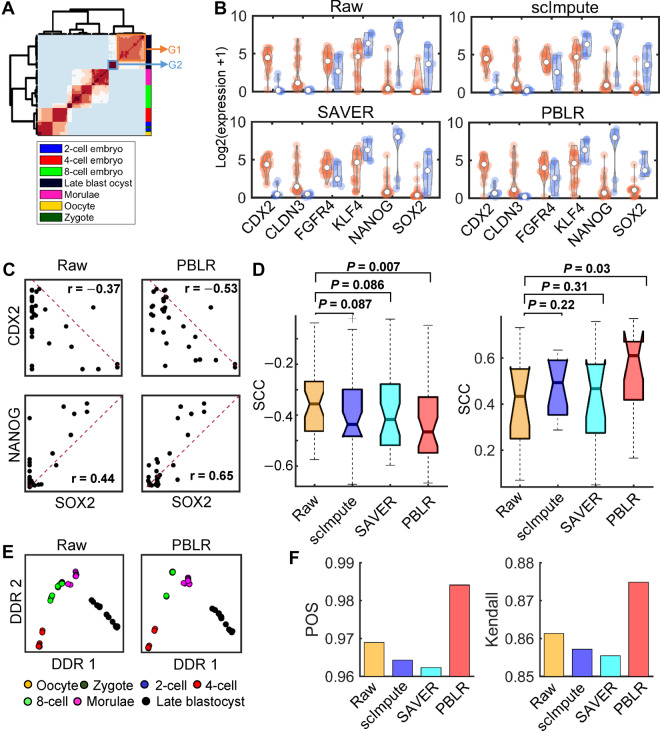
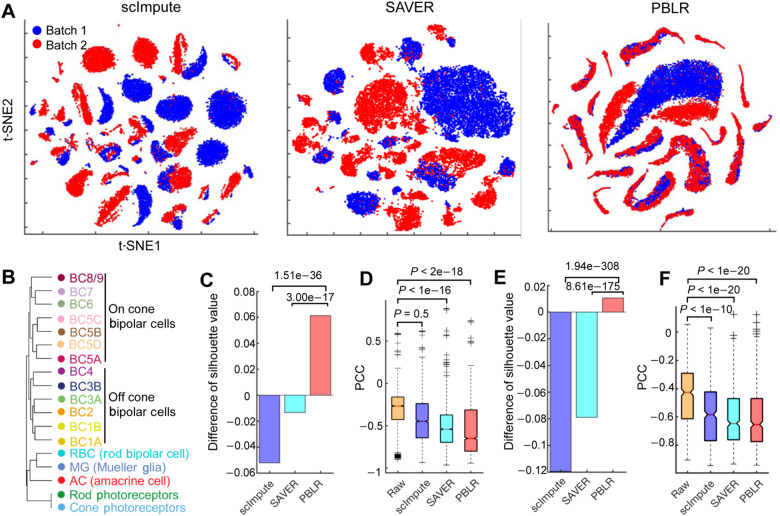
Single-cell RNA sequencing (scRNA-seq) provides a powerful tool to determine expression patterns of thousands of individual cells. However, the analysis of scRNA-seq data remains a computational challenge due to the high technical noise such as the presence of dropout events that lead to a large proportion of zeros for expressed genes. Taking into account the cell heterogeneity and the relationship between dropout rate and expected expression level, we present a cell sub-population based bounded low-rank (PBLR) method to impute the dropouts of scRNA-seq data. Through application to both simulated and real scRNA-seq datasets, PBLR is shown to be effective in recovering dropout events, and it can dramatically improve the low-dimensional representation and the recovery of gene‒gene relationships masked by dropout events compared to several state-of-the-art methods. Moreover, PBLR also detects accurate and robust cell sub-populations automatically, shedding light on its flexibility and generality for scRNA-seq data analysis.
单细胞 RNA 测序 (scRNA-seq) 提供了一种强大的工具,可以确定数千个单个细胞的表达模式。然而,由于技术噪声高,如存在缺失事件,导致表达基因的大量零值,因此 scRNA-seq 数据的分析仍然是一个计算挑战。考虑到细胞异质性以及缺失率和预期表达水平之间的关系,我们提出了一种基于细胞亚群的有界低秩 (PBLR) 方法来推断 scRNA-seq 数据的缺失值。通过对模拟和真实 scRNA-seq 数据集的应用,PBLR 被证明在恢复缺失事件方面非常有效,与几种最先进的方法相比,它可以显著改善由缺失事件掩盖的低维表示和基因-基因关系的恢复。此外,PBLR 还可以自动检测准确和稳健的细胞亚群,这表明它在 scRNA-seq 数据分析方面具有灵活性和通用性。