Han Ranran, Wan Jieru, Han Xiaoning, Ren Honglei, Falck John R, Munnuri Sailu, Yang Zeng-Jin, Koehler Raymond C
Department of Anesthesiology and Critical Care Medicine, The Johns Hopkins University, Baltimore, MD, United States.
Department of Biochemistry, University of Texas Southwestern Medical Center, Dallas, TX, United States.
Front Neurol. 2021 Nov 12;12:763419. doi: 10.3389/fneur.2021.763419. eCollection 2021.
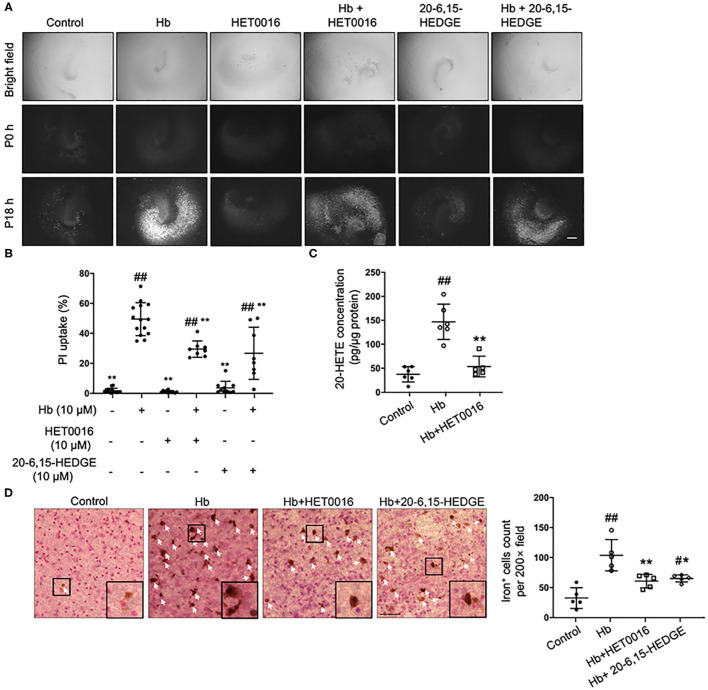
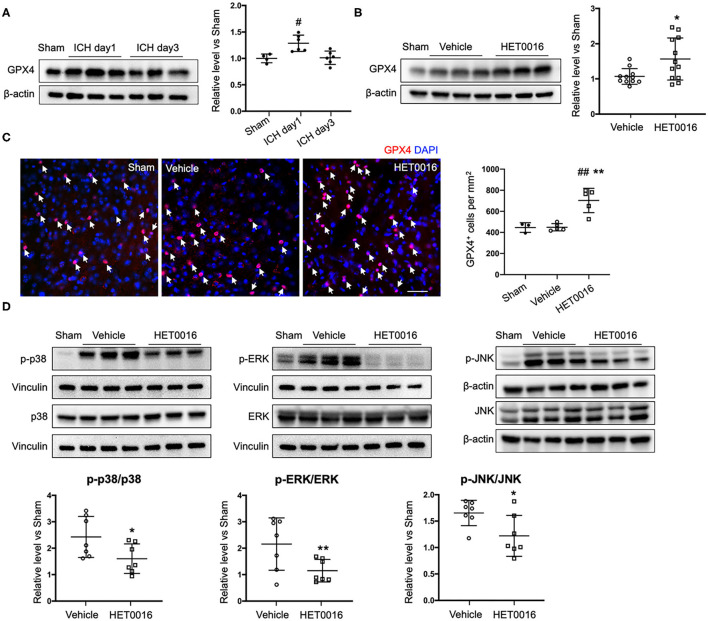
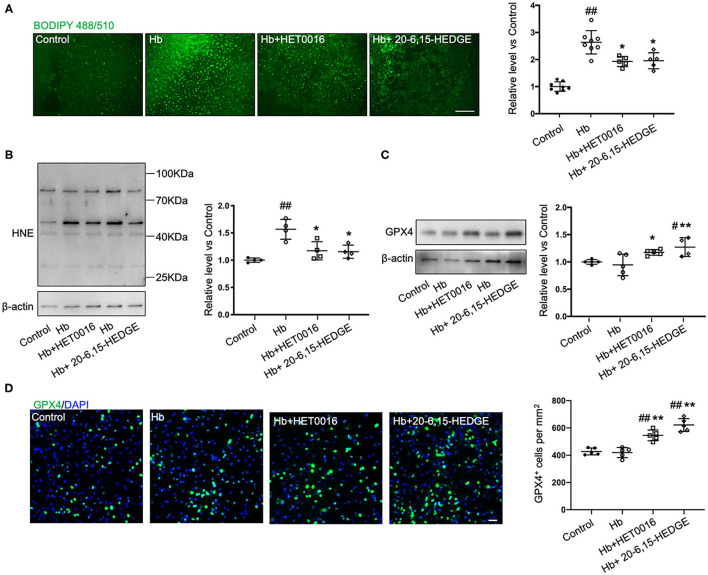
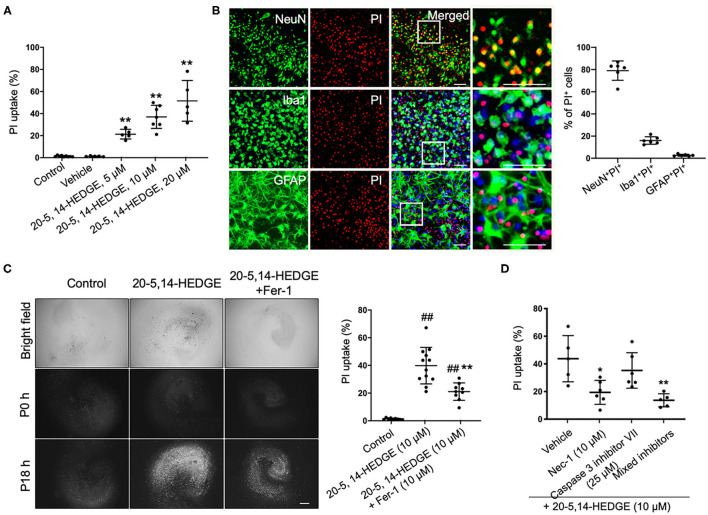
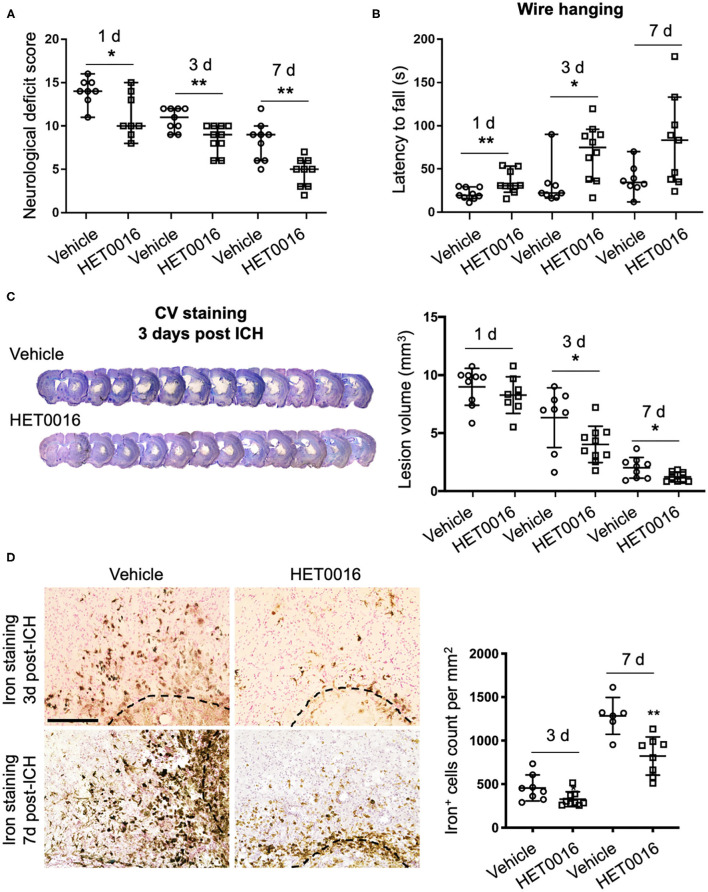
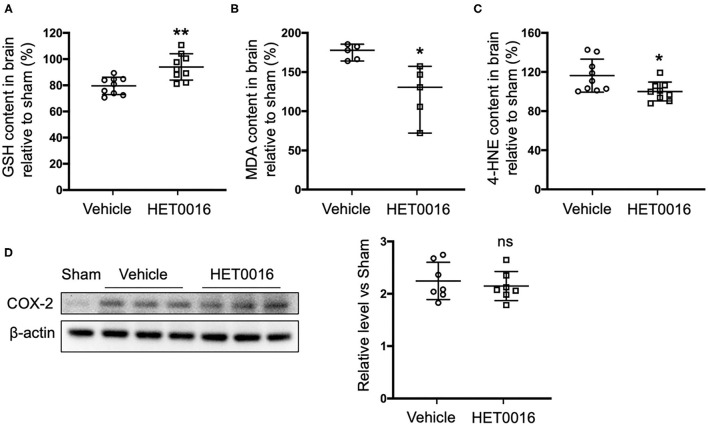
Intracerebral hemorrhage (ICH) is a highly fatal type of stroke that leads to various types of neuronal death. Recently, ferroptosis, a form of cell death resulting from iron-dependent lipid peroxide accumulation, was observed in a mouse ICH model. N-hydroxy-N'-(4-n-butyl-2-methylphenyl)-formamidine (HET0016), which inhibits synthesis of the arachidonic acid metabolite 20-hydroxyeicosatetraenoic acid (20-HETE), has shown a protective effect after ICH. However, the underlying mechanisms of the neuroprotective effect need further investigation. We explored whether 20-HETE participates in ICH-induced ferroptosis by using hemoglobin-treated organotypic hippocampal slice cultures (OHSCs) and by using a collagenase-induced ICH mouse model. , we found that the 20-HETE synthesis inhibitor HET0016 and antagonist 20-6,15-HEDGE reduced hemoglobin-induced cell death, iron deposition, and lipid reactive oxygen species levels in OHSCs. Furthermore, 20-HETE inhibition in OHSCs increased the expression of glutathione peroxidase (GPX) 4, an antioxidant enzyme that serves as a main regulator of ferroptosis. In contrast, exposure of OHSCs to the 20-HETE stable mimetic 20-5,14-HEDGE induced cell death that was significantly inhibited by the ferroptosis inhibitor ferrostatin-1. , HET0016 treatment ameliorated focal deficits, reduced lesion volume, and decreased iron accumulation around the lesion at day 3 and 7 after ICH. In addition, lipid peroxidation was decreased and expression of GPX4 was increased in the HET0016-treated ICH group. The mitogen-activated protein kinase pathway also was inhibited by HET0016 . These results indicate that 20-HETE contributes to ICH-induced acute brain injury in part by activating ferroptosis pathways, thereby providing an upstream target for inhibiting ferroptosis.
脑出血(ICH)是一种致死率很高的中风类型,可导致多种类型的神经元死亡。最近,在小鼠脑出血模型中观察到铁死亡,这是一种由于铁依赖性脂质过氧化物积累而导致的细胞死亡形式。N-羟基-N'-(4-正丁基-2-甲基苯基)甲脒(HET0016)可抑制花生四烯酸代谢物20-羟基二十碳四烯酸(20-HETE)的合成,在脑出血后显示出保护作用。然而,这种神经保护作用的潜在机制需要进一步研究。我们通过使用血红蛋白处理的器官型海马脑片培养物(OHSCs)以及胶原酶诱导的脑出血小鼠模型,探讨了20-HETE是否参与脑出血诱导的铁死亡。我们发现,20-HETE合成抑制剂HET0016和拮抗剂20-6,15-HEDGE可减少血红蛋白诱导的OHSCs中的细胞死亡、铁沉积和脂质活性氧水平。此外,在OHSCs中抑制20-HETE可增加谷胱甘肽过氧化物酶(GPX)4的表达,GPX4是一种抗氧化酶,是铁死亡的主要调节因子。相反,将OHSCs暴露于20-HETE稳定模拟物20-5,14-HEDGE可诱导细胞死亡,铁死亡抑制剂铁抑素-1可显著抑制这种死亡。HET0016治疗可改善局灶性缺陷,减少病变体积,并在脑出血后第3天和第7天减少病变周围的铁积累。此外,在HET0016治疗的脑出血组中,脂质过氧化减少,GPX4表达增加。丝裂原活化蛋白激酶途径也被HET0016抑制。这些结果表明,20-HETE部分通过激活铁死亡途径导致脑出血诱导的急性脑损伤,从而为抑制铁死亡提供了一个上游靶点。