Vollrath Paul, Chawla Harmeet S, Alnajar Dima, Gabur Iulian, Lee HueyTyng, Weber Sven, Ehrig Lennard, Koopmann Birger, Snowdon Rod J, Obermeier Christian
Department of Plant Breeding, IFZ Research Centre for Biosystems, Land Use and Nutrition, Justus Liebig University Giessen, Giessen, Germany.
Department of Plant Sciences, Crop Development Centre, University of Saskatchewan, Saskatoon, SK, Canada.
Front Plant Sci. 2021 Nov 18;12:749491. doi: 10.3389/fpls.2021.749491. eCollection 2021.
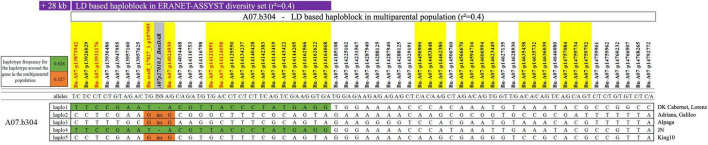
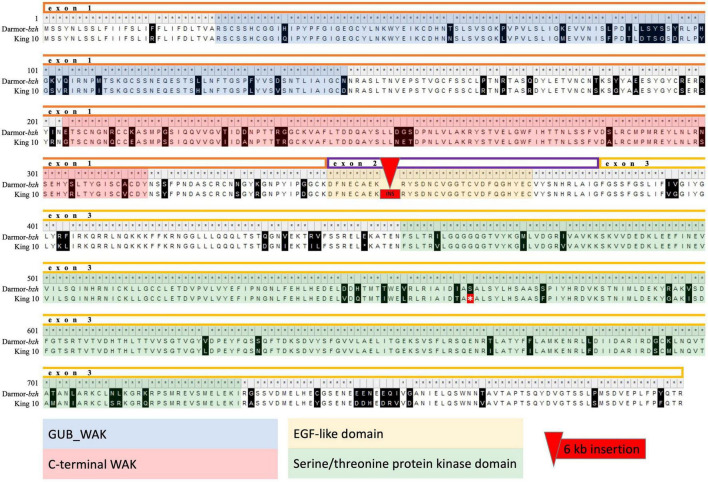
Blackleg is one of the major fungal diseases in oilseed rape/canola worldwide. Most commercial cultivars carry gene-mediated qualitative resistances that confer a high level of race-specific protection against , the causal fungus of blackleg disease. However, monogenic resistances of this kind can potentially be rapidly overcome by mutations in the pathogen's avirulence genes. To counteract pathogen adaptation in this evolutionary arms race, there is a tremendous demand for quantitative background resistance to enhance durability and efficacy of blackleg resistance in oilseed rape. In this study, we characterized genomic regions contributing to quantitative resistance by genome-wide association studies in a multiparental mapping population derived from six parental elite varieties exhibiting quantitative resistance, which were all crossed to one common susceptible parental elite variety. Resistance was screened using a fungal isolate with no corresponding avirulence () to major genes present in the parents of the mapping population. Genome-wide association studies revealed eight significantly associated quantitative trait loci (QTL) on chromosomes A07 and A09, with small effects explaining 3-6% of the phenotypic variance. Unexpectedly, the qualitative blackleg resistance gene was found to be located within a resistance-associated haploblock on chromosome A07. Furthermore, long-range sequence data spanning this haploblock revealed high levels of single-nucleotide and structural variants within the coding sequence among the parents of the mapping population. The results suggest that novel variants of could play a previously unknown role in expression of quantitative disease resistance in oilseed rape.
黑胫病是全球油菜籽/双低油菜的主要真菌病害之一。大多数商业栽培品种携带基因介导的定性抗性,可对黑胫病的致病真菌 提供高水平的小种特异性保护。然而,这种单基因抗性可能会因病原体无毒基因的突变而迅速被克服。为了在这场进化的军备竞赛中对抗病原体的适应性,迫切需要定量背景抗性来提高油菜黑胫病抗性的持久性和有效性。在本研究中,我们通过全基因组关联研究,在一个多亲本作图群体中鉴定了有助于定量 抗性的基因组区域,该群体由六个表现出定量抗性的亲本优良品种衍生而来,这些品种均与一个共同的感病亲本优良品种杂交。使用对作图群体亲本中存在的主要 基因无相应无毒()的真菌分离株筛选抗性。全基因组关联研究在A07和A09染色体上揭示了八个显著相关的数量性状位点(QTL),其效应较小,解释了3-6%的表型变异。出乎意料的是,定性黑胫病抗性基因 被发现位于A07染色体上一个与抗性相关的单倍体区域内。此外,跨越该单倍体区域的长程序列数据显示,作图群体亲本中 编码序列内存在高水平的单核苷酸和结构变异。结果表明, 的新变体可能在油菜定量抗病性表达中发挥了以前未知的作用。