Department of Neurology, Washington University School of Medicine in St. Louis, St. Louis, MO, USA.
Department of Psychology, Ashoka University, Rai, India.
J Neuroinflammation. 2021 Dec 11;18(1):289. doi: 10.1186/s12974-021-02322-9.
Current therapies targeting several neurotransmitter systems are only able to partially mitigate the symptoms of stress- and trauma-related disorder. Stress and trauma-related disorders lead to a prominent inflammatory response in humans, and in pre-clinical models. However, mechanisms underlying the induction of neuroinflammatory response in PTSD and anxiety disorders are not clearly understood. The present study investigated the mechanism underlying the activation of proinflammatory NLRP3 inflammasome and IL1β in mouse models of stress.
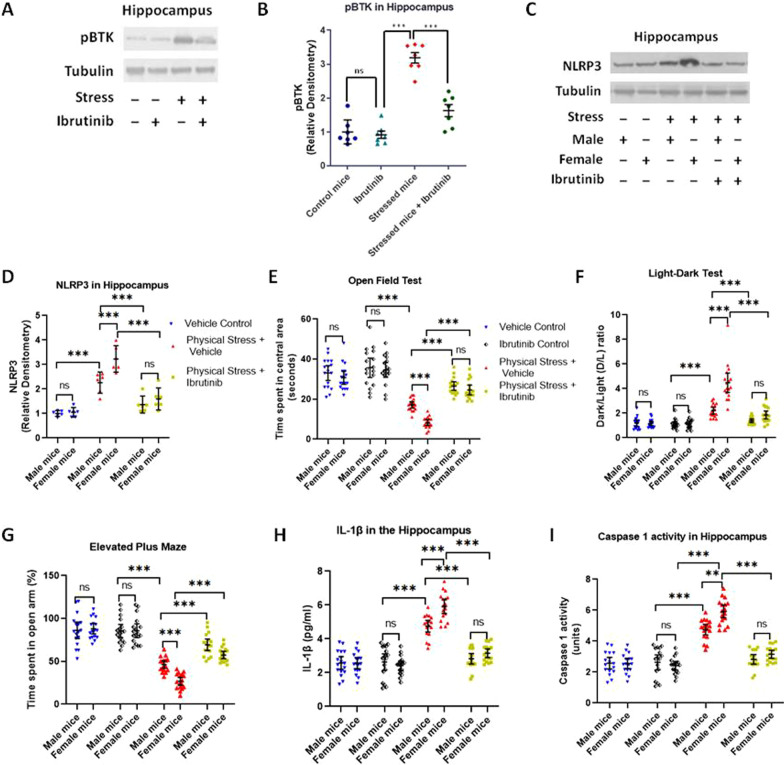
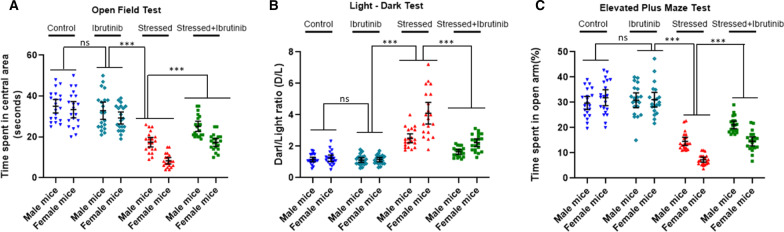
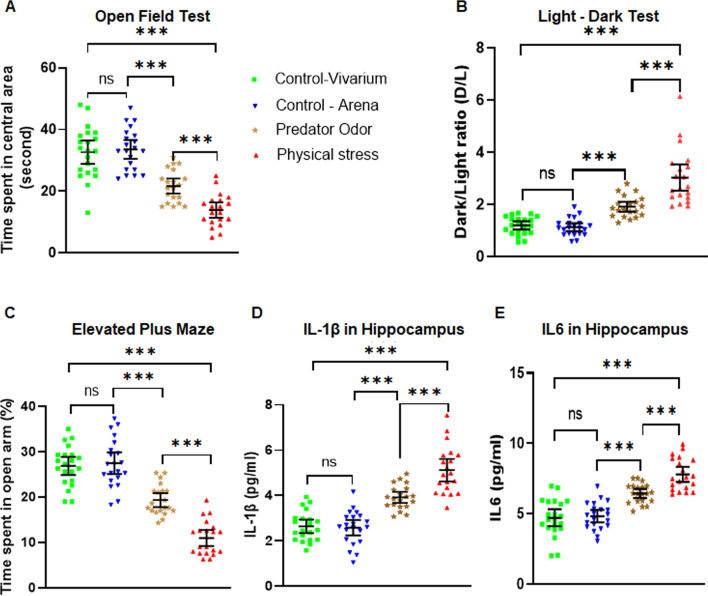
We used two mouse models of stress, i.e., mice subjected to physical restraint stress with brief underwater submersion, and predator odor stress. Mice were injected with MCC950, a small molecule specific inhibitor of NLRP3 activation. To pharmacologically inhibit BTK, a specific inhibitor ibrutinib was used. To validate the observation from ibrutinib studies, a separate group of mice was injected with another BTK-specific inhibitor LFM-A13. Seven days after the induction of stress, mice were examined for anxious behavior using open field test (OFT), light-dark test (LDT), and elevated plus maze test (EPM). Following the behavior tests, hippocampus and amygdale were extracted and analyzed for various components of NLRP3-caspase 1-IL1β pathway. Plasma and peripheral blood mononuclear cells were also used to assess the induction of NLRP3-Caspase 1-IL-1β pathway in stressed mice.
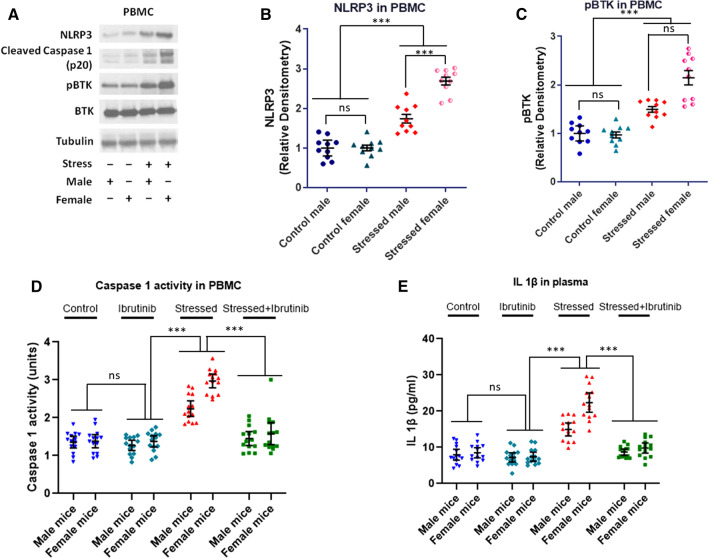
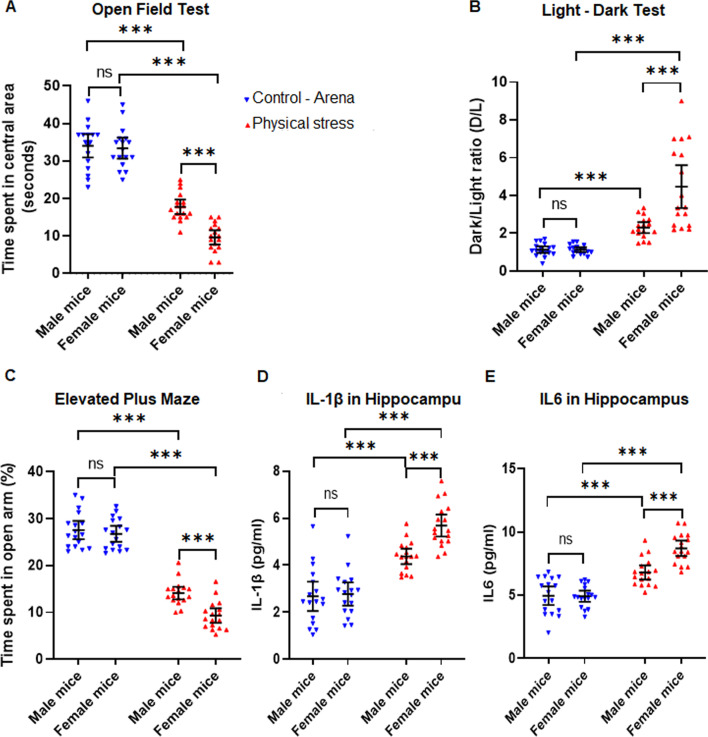
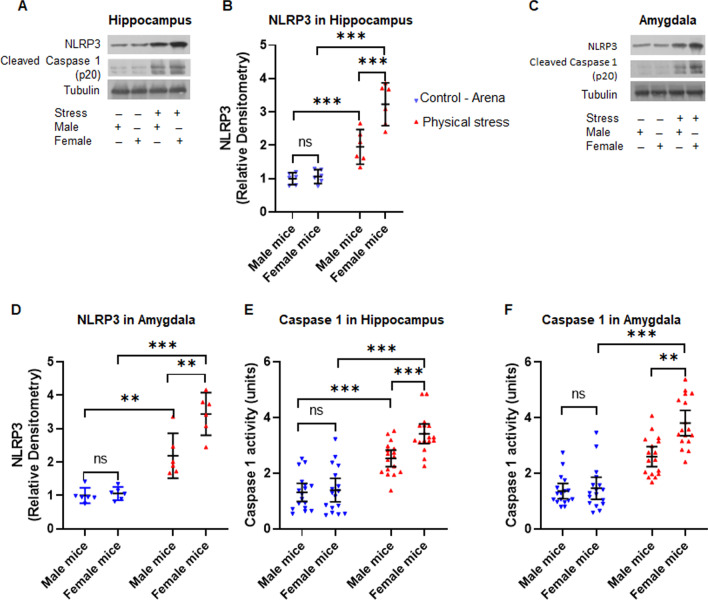
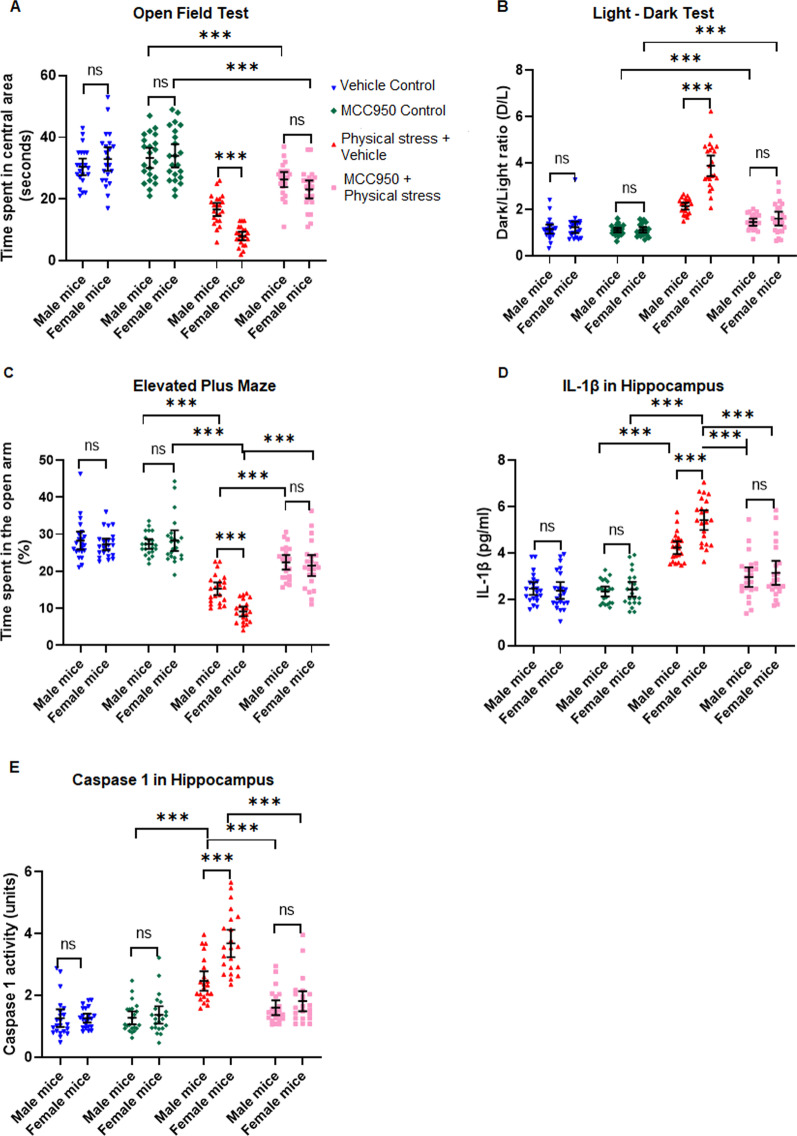
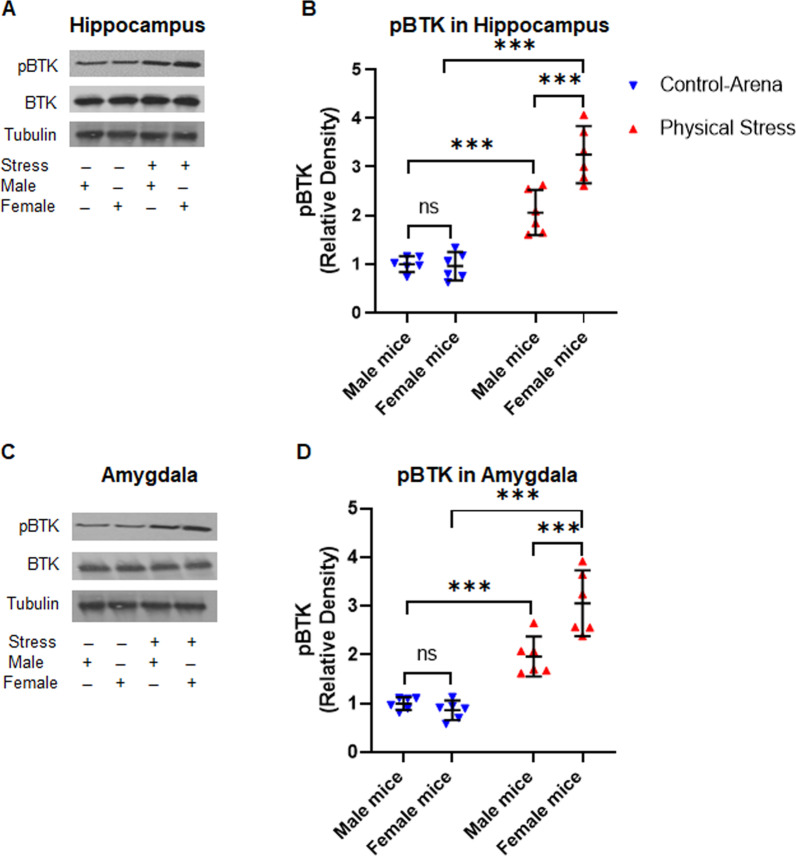
Using two different pre-clinical models of stress, we demonstrate heightened anxious behavior in female mice as compared to their male counterparts. Stressed animals exhibited upregulation of proinflammatory IL1β, IL-6, Caspase 1 activity and NLRP3 inflammasome activation in brain, which were significantly higher in female mice. Pharmacological inhibition of NLRP3 inflammasome activation led to anxiolysis as well as attenuated neuroinflammatory response. Further, we observed induction of activated Bruton's tyrosine kinase (BTK), an upstream positive-regulator of NLRP3 inflammasome activation, in hippocampus and amygdala of stressed mice. Next, we conducted proof-of-concept pharmacological BTK inhibitor studies with ibrutinib and LFM-A13. In both sets of experiments, we found BTK inhibition led to anxiolysis and attenuated neuroinflammation, as indicated by significant reduction of NLRP3 inflammasome and proinflammatory IL-1β in hippocampus and amygdala. Analysis of plasma and peripheral blood mononuclear cells indicated peripheral induction of NLRP3-caspase 1-IL1β pathway in stressed mice.
Our study identified BTK as a key upstream regulator of neuroinflammation, which drives anxiogenic behavior in mouse model of stress. Further, we demonstrated the sexually divergent activation of BTK, providing a clue to heightened neuroinflammation and anxiogenic response to stress in females as compared to their male counterparts. Our data from the pharmacological inhibition studies suggest BTK as a novel target for the development of potential clinical treatment of PTSD and anxiety disorders. Induction of pBTK and NLRP3 in peripheral blood mononuclear cells of stressed mice suggest the potential effect of stress on systemic inflammation.
目前针对多种神经递质系统的治疗方法只能部分缓解与应激和创伤相关的障碍的症状。应激和创伤相关障碍会导致人类和临床前模型中出现明显的炎症反应。然而, PTSD 和焦虑障碍中神经炎症反应诱导的机制尚不清楚。本研究探讨了应激诱导的促炎 NLRP3 炎性小体和 IL1β 在小鼠模型中的激活机制。
我们使用了两种应激的小鼠模型,即经历短暂水下浸泡的身体束缚应激的小鼠和捕食者气味应激的小鼠。给小鼠注射 NLRP3 激活的小分子特异性抑制剂 MCC950。为了抑制 BTK 的药理作用,使用了一种特异性抑制剂 ibrutinib。为了验证 ibrutinib 研究的观察结果,另一组小鼠注射了另一种 BTK 特异性抑制剂 LFM-A13。应激诱导 7 天后,使用旷场试验 (OFT)、明暗试验 (LDT) 和高架十字迷宫试验 (EPM) 检查小鼠的焦虑行为。行为测试后,提取海马体和杏仁核并分析 NLRP3-caspase 1-IL1β 途径的各种成分。还使用血浆和外周血单核细胞来评估应激小鼠中 NLRP3-Caspase 1-IL-1β 途径的诱导。
使用两种不同的应激临床前模型,我们证明与雄性小鼠相比,雌性小鼠表现出更高的焦虑行为。应激动物表现出促炎细胞因子 IL1β、IL-6、Caspase 1 活性和大脑中的 NLRP3 炎性小体激活增加,而雌性小鼠的这些增加更为明显。NLRP3 炎性小体激活的药理学抑制导致焦虑减轻和神经炎症反应减弱。此外,我们观察到应激小鼠海马体和杏仁核中激活的 Bruton 酪氨酸激酶 (BTK),BTK 是 NLRP3 炎性小体激活的上游正调节剂。接下来,我们进行了使用 ibrutinib 和 LFM-A13 的 BTK 抑制剂的概念验证药理学研究。在这两组实验中,我们发现 BTK 抑制导致焦虑减轻和神经炎症减弱,这表明 NLRP3 炎性小体和海马体和杏仁核中的促炎 IL-1β 显著减少。对血浆和外周血单核细胞的分析表明,应激小鼠的外周血中 NLRP3-caspase 1-IL1β 途径被诱导。
我们的研究确定了 BTK 作为神经炎症的关键上游调节剂,它在应激的小鼠模型中驱动焦虑行为。此外,我们证明了 BTK 的性别差异激活,这为与应激相关的女性与男性相比,神经炎症和焦虑反应增强提供了线索。我们的药理学抑制研究数据表明 BTK 是 PTSD 和焦虑障碍潜在临床治疗的新靶点。应激小鼠外周血单核细胞中 pBTK 和 NLRP3 的诱导表明应激对全身炎症的潜在影响。