Dehani Mohammad, Zare-Abdollahi Davood, Bushehri Ata, Dehghani Azadeh, Effati Jalil, Miratashi Seyed Ali Mohammad, Khorram Khorshid Hamid Reza
Genetics Research Center, University of Social Welfare and Rehabilitation Sciences, Tehran, Iran.
Fetal Health Research Center, Hope Generation Foundation, Tehran, Iran.
Avicenna J Med Biotechnol. 2021 Oct-Dec;13(4):230-233.
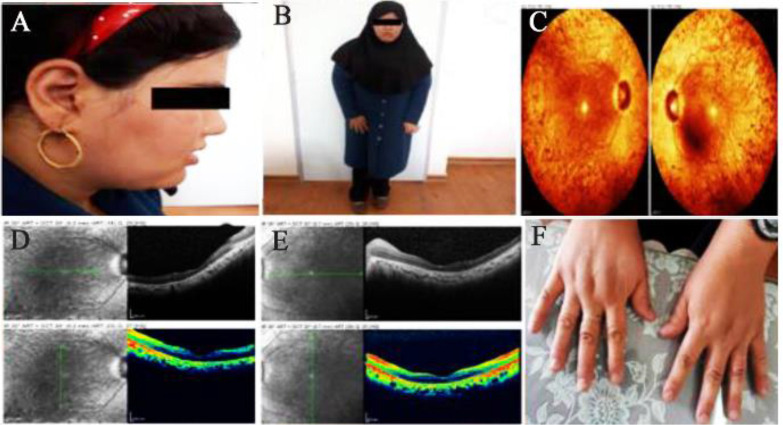
Bardet-Biedl Syndrome (BBS) is a rare pleiotropic autosomal recessive disease related to ciliopathies with approximately 25 causative genes. BBS is a multisystemic disorder with wide spectrum of manifestations including truncal obesity, retinal dystrophy, male hypogenitalism, postaxial polydactyly, learning difficulties, and renal abnormalities.
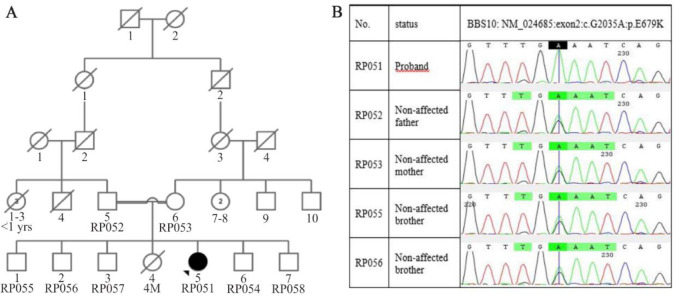
A consanguineous Iranian family with a 28-year-old daughter affected with BBS, resulting from a first cousin marriage, was examined. After clinical examination, Whole Exome Sequencing (WES) was applied. Following the analysis of exome data, Sanger sequencing was used to confirm as well as to co-segregate the candidate variant with the phenotype.
A novel homozygous variant [c. 2035G>A (p.E679K)] in exon 2 of the gene was found which was categorized as likely pathogenic based on American College of Medical Genetics and Genomics (ACMG) guidelines and criteria. In this study, the variant was fully co-segregated with the phenotype in the family.
Despite overlapping with other ciliopathies in terms of the phenotype, the BBS has high genetic heterogeneity and clinical variability even among affected members of a family. The symptoms observed in patients are largely related to the genes involved and the type of mutations in the BBS. In this study, in addition to phenotype description of the proband harboring a novel disease-causing variant in gene, the spectrum of BBS symptoms was expanded. The findings of this study can be useful in genetic counseling, especially for risk estimation and prenatal diagnosis.
巴德-比埃尔综合征(BBS)是一种罕见的多效性常染色体隐性疾病,与纤毛病相关,约有25个致病基因。BBS是一种多系统疾病,表现形式广泛,包括躯干肥胖、视网膜营养不良、男性生殖器发育不全、轴后多指畸形、学习困难和肾脏异常。
对一个近亲结婚的伊朗家庭进行了检查,该家庭中一名28岁的女儿患有BBS,系表亲结婚所致。临床检查后,应用了全外显子组测序(WES)。在分析外显子组数据后,使用桑格测序来确认候选变异并将其与表型进行共分离。
在该基因的第2外显子中发现了一个新的纯合变异[c. 2035G>A(p.E679K)],根据美国医学遗传学与基因组学学会(ACMG)的指南和标准,该变异被归类为可能致病。在本研究中,该变异与家族中的表型完全共分离。
尽管BBS在表型上与其他纤毛病有重叠,但即使在一个家族的受影响成员中,BBS也具有高度的遗传异质性和临床变异性。患者观察到的症状很大程度上与所涉及的基因以及BBS中的突变类型有关。在本研究中,除了对携带该基因新致病变异的先证者进行表型描述外,还扩展了BBS症状谱。本研究结果可用于遗传咨询,特别是风险评估和产前诊断。