Diagnostic and Research Center for Molecular BioMedicine, Medical University of Graz, Graz, Austria.
Department of Medical Genetics, Kasturba Medical College, Manipal, Manipal Academy of Higher Education, Manipal, India.
J Med Genet. 2022 Oct;59(10):957-964. doi: 10.1136/jmedgenet-2021-108061. Epub 2021 Dec 16.
Mucopolysaccharidoses (MPS) are monogenic metabolic disorders that significantly affect the skeleton. Eleven enzyme defects in the lysosomal degradation of glycosaminoglycans (GAGs) have been assigned to the known MPS subtypes (I-IX). Arylsulfatase K (ARSK) is a recently characterised lysosomal hydrolase involved in GAG degradation that removes the 2-O-sulfate group from 2-sulfoglucuronate. Knockout of in mice was consistent with mild storage pathology, but no human phenotype has yet been described.
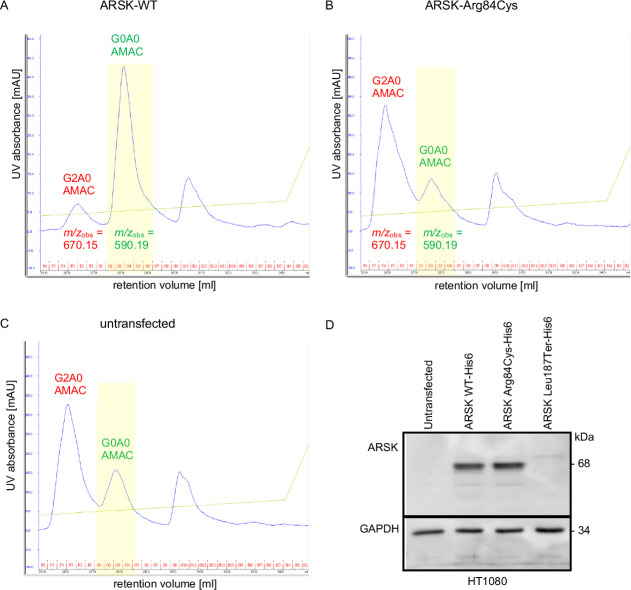
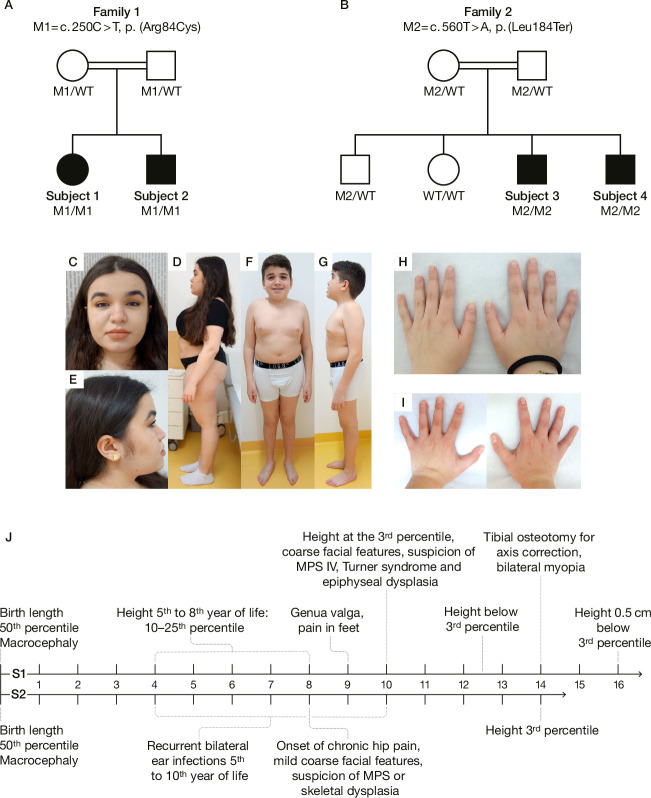
In this study, we report four affected individuals of two unrelated consanguineous families with homozygous variants c.250C>T, p.(Arg84Cys) and c.560T>A, p.(Leu187Ter) in , respectively. Functional consequences of the two variants were assessed by mutation-specific constructs derived by site-directed mutagenesis, which were ectopically expressed in HT1080 cells. Urinary GAG excretion was analysed by dimethylene blue and electrophoresis, as well as liquid chromatography/mass spectrometry (LC-MS)/MS analysis.
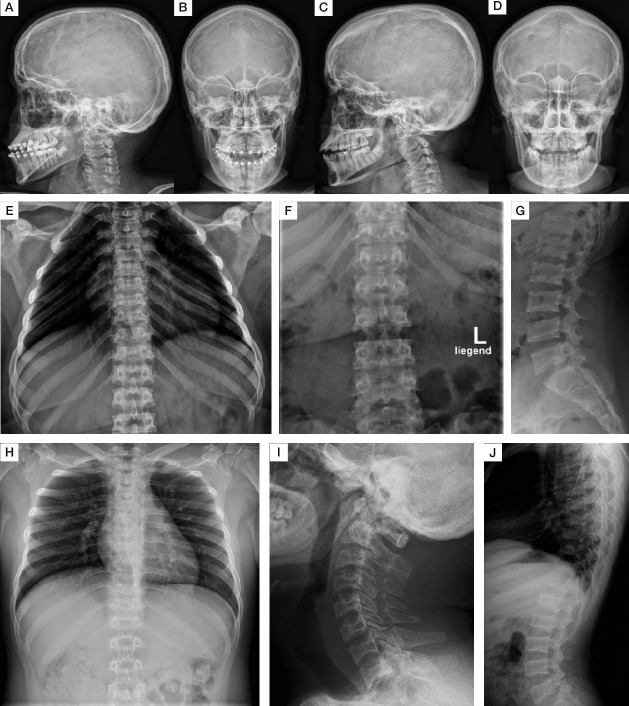
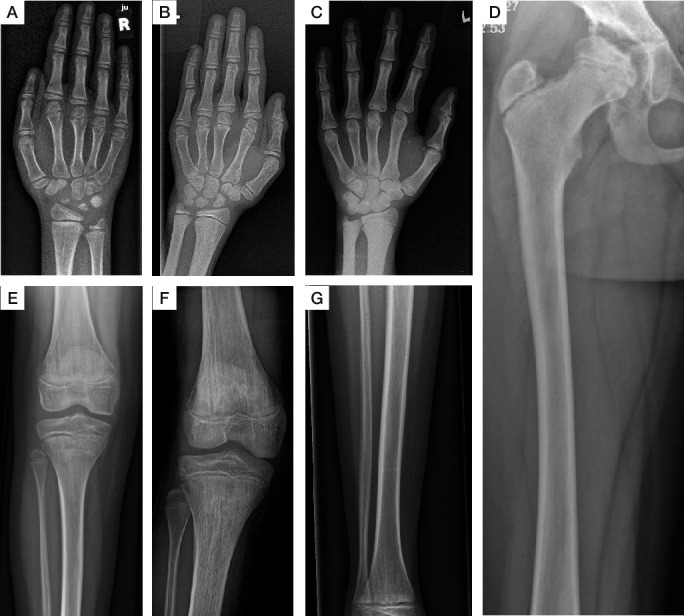
The phenotypes of the affected individuals include MPS features, such as short stature, coarse facial features and dysostosis multiplex. Reverse phenotyping in two of the four individuals revealed additional cardiac and ophthalmological abnormalities. Mild elevation of dermatan sulfate was detected in the two subjects investigated by LC-MS/MS. Human HT1080 cells expressing the ARSK-Leu187Ter construct exhibited absent protein levels by western blot, and cells with the ARSK-Arg84Cys construct showed markedly reduced enzyme activity in an ARSK-specific enzymatic assay against 2-O-sulfoglucuronate-containing disaccharides as analysed by C18-reversed-phase chromatography followed by MS.
Our work provides a detailed clinical and molecular characterisation of a novel subtype of mucopolysaccharidosis, which we suggest to designate subtype X.
黏多糖贮积症(MPS)是一种单基因代谢紊乱疾病,严重影响骨骼。已知 MPS 亚型(I-IX)共有 11 种溶酶体降解糖胺聚糖(GAG)的酶缺陷。芳基硫酸酯酶 K(ARSK)是一种新发现的溶酶体水解酶,参与 GAG 降解,可从 2-磺基葡萄糖醛酸中去除 2-O-硫酸基团。小鼠的基因敲除与轻度贮存病理学一致,但尚未描述人类表型。
本研究报告了两个无血缘关系的近亲家庭的四个受影响个体,他们分别携带 ARSK 基因中的 c.250C>T,p.(Arg84Cys)和 c.560T>A,p.(Leu187Ter)纯合变体。通过定点诱变获得突变特异性 ARSK 构建体,异位表达于 HT1080 细胞,评估了这两种 ARSK 变体的功能后果。通过二甲亚砜和电泳以及液相色谱/质谱(LC-MS)/MS 分析来分析尿 GAG 排泄。
受影响个体的表型包括 MPS 特征,如身材矮小、面容粗糙和多发性骨发育不良。在四个受影响个体中的两个个体的反向表型研究中发现了其他心脏和眼科异常。通过 LC-MS/MS 分析,在两个被研究的个体中检测到轻微升高的硫酸皮肤素。用 ARSK-Leu187Ter 构建体表达的 HT1080 细胞通过 Western blot 显示蛋白水平缺失,而用 ARSK-Arg84Cys 构建体表达的细胞在 ARSK 特异性酶分析中对含 2-O-磺基葡萄糖醛酸的二糖的酶活性明显降低,通过 C18 反相色谱法分析后用 MS 检测。
我们的工作提供了一种新型黏多糖贮积症亚型的详细临床和分子特征,我们建议将其命名为 X 型。