Department of Pediatrics, Graduate School of Medical Sciences, Kumamoto University, 1-1-1 Honjo, Chuo-ku, Kumamoto City, Kumamoto, 860-8556, Japan.
Kumamoto-Ashikita Medical Center for Disabled Children, Kumamoto, Japan.
Orphanet J Rare Dis. 2021 Dec 18;16(1):516. doi: 10.1186/s13023-021-02146-z.
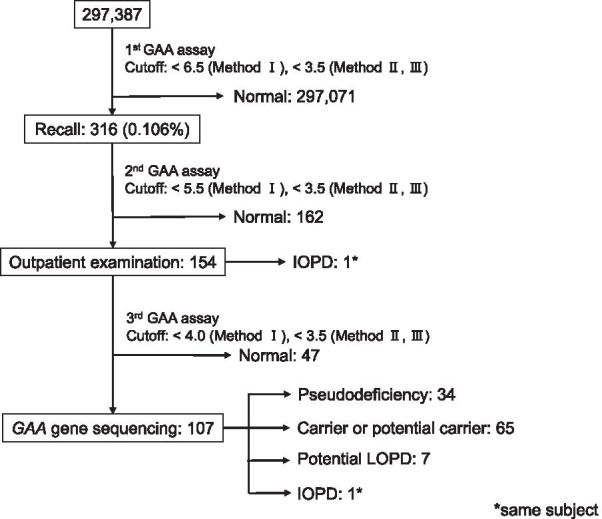
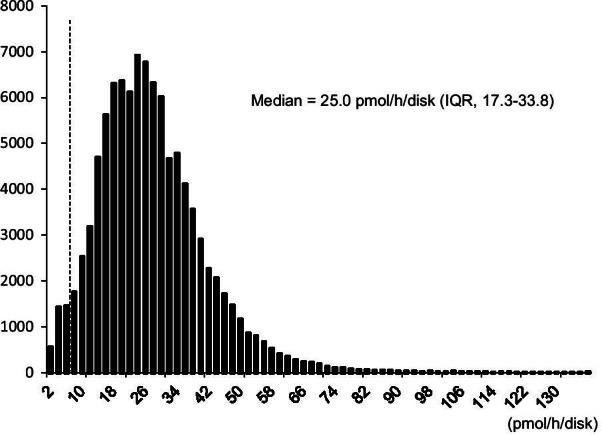
Pompe disease is an autosomal recessive inherited metabolic disorder caused by a deficiency of the acid α-glucosidase (GAA). Pompe disease manifests as an accumulation of lysosomal glycogen in the skeletal and heart muscle. We conducted newborn screening (NBS) for Pompe disease in Japan from April 2013 to October 2020 to determine the feasibility and utility of NBS for Pompe disease.
From the 296,759 newborns whose enzyme activity was measured, 107 of which underwent GAA analysis, we found one patient with infantile-onset Pompe disease (IOPD) and seven with potential late-onset Pompe disease (LOPD). We identified 34 pseudodeficient individuals and 65 carriers or potential carriers. The frequency of patients with IOPD was similar to that in the United States, but significantly lower than that in Taiwan. One patient with IOPD underwent early enzyme replacement therapy within a month after birth before presenting exacerbated manifestations, whereas those with potential LOPD showed no manifestations during the follow-up period of six years.
The frequency of IOPD in Japan was similar to that in the United States, where NBS for Pompe disease is recommended. This indicates that NBS for Pompe disease may also be useful in Japan. Therefore, it should be used over a wider region in Japan.
庞贝病是一种常染色体隐性遗传代谢疾病,由酸性α-葡萄糖苷酶(GAA)缺乏引起。庞贝病表现为溶酶体糖原在骨骼肌和心肌中的积累。我们于 2013 年 4 月至 2020 年 10 月在日本进行了庞贝病的新生儿筛查(NBS),以确定 NBS 对庞贝病的可行性和实用性。
在对 296759 名新生儿进行酶活性测量的基础上,我们对其中的 107 名新生儿进行了 GAA 分析,发现了一名婴儿型庞贝病(IOPD)患者和七名潜在晚发型庞贝病(LOPD)患者。我们还鉴定了 34 名假缺陷个体和 65 名携带者或潜在携带者。IOPD 患者的频率与美国相似,但明显低于中国台湾。一名 IOPD 患者在出现恶化表现之前,于出生后一个月内接受了早期酶替代治疗,而潜在的 LOPD 患者在六年的随访期间没有出现任何症状。
日本的 IOPD 频率与美国相似,美国推荐对庞贝病进行 NBS。这表明 NBS 对庞贝病可能在日本也同样有用。因此,它应该在日本更大的地区使用。