Dept of Thoracic Medicine, The Prince Charles Hospital, Brisbane, Australia
Interstitial Lung Disease Unit, Royal Brompton Hospital, London, UK.
Eur Respir Rev. 2021 Dec 22;30(162). doi: 10.1183/16000617.0177-2021. Print 2021 Dec 31.
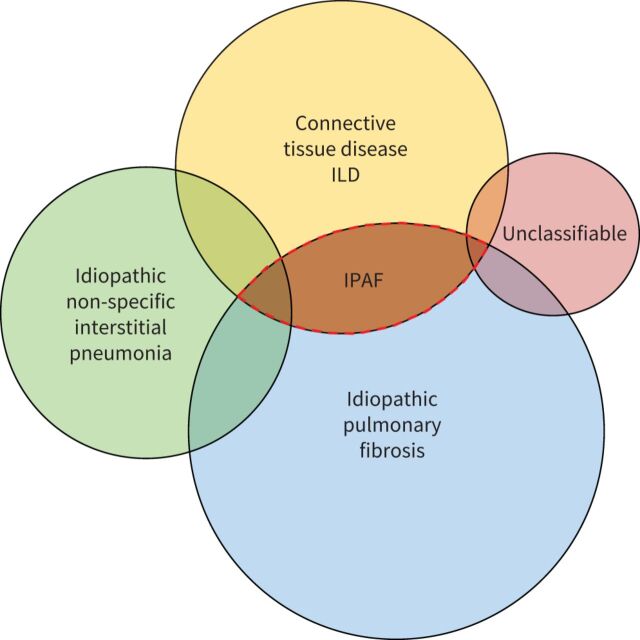
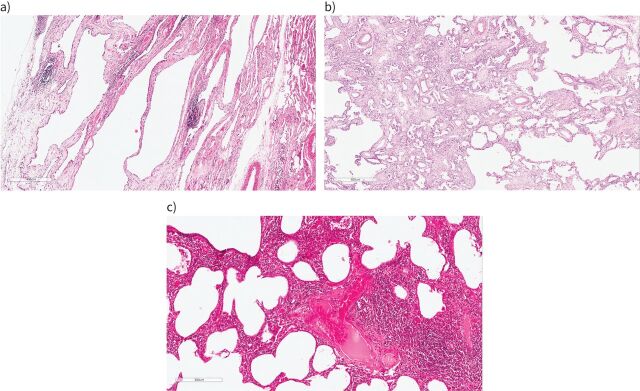
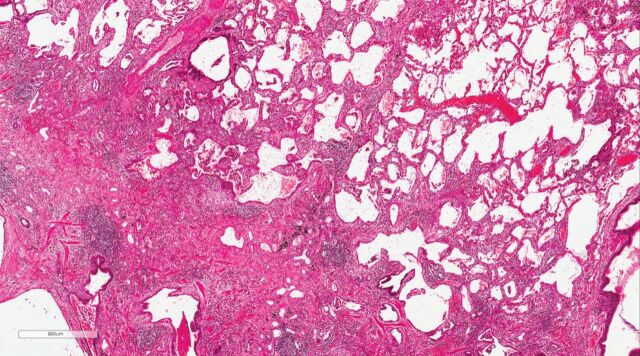
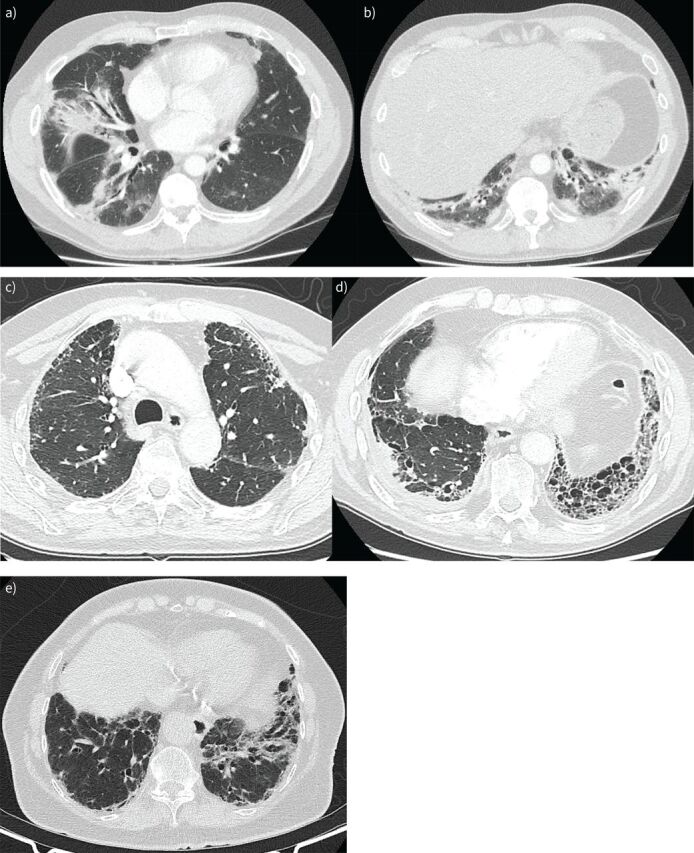
The presence of clinical, serological and/or radiological features suggestive, but not confirmatory, of a defined connective tissue disease in patients with interstitial lung disease is a relatively frequent occurrence. In 2015, the European Respiratory Society and the American Thoracic Society proposed classification criteria for the interstitial pneumonia with autoimmune features (IPAF) research entity to capture such patients in a standardised manner, with the intention of nurturing clinical research. This initiative resulted in the publication of several series of IPAF patients, with significant variation between cohorts in clinical characteristics, outcome and the application of IPAF criteria in patient selection. From this increasing body of published work, it has become apparent that revision of IPAF criteria is now required in order to justify the eventual designation of IPAF as a standalone diagnostic term, as opposed to a provisional entity put forward as a basis for clinical research. This review covers the current state of IPAF, conclusions that can and cannot be drawn from the IPAF evidence base, and ongoing uncertainties that require further expert group consideration.
在患有间质性肺疾病的患者中,存在临床、血清学和/或影像学特征提示,但不足以确诊的特定结缔组织病,这种情况较为常见。2015 年,欧洲呼吸学会和美国胸科学会提出了自身免疫特征性间质性肺炎(IPAF)研究实体的分类标准,旨在以标准化的方式对这些患者进行分类,以促进临床研究。这一举措促成了一系列 IPAF 患者的发表,不同队列之间在临床特征、结局以及 IPAF 标准在患者选择中的应用方面存在显著差异。从越来越多的已发表工作中可以明显看出,现在需要对 IPAF 标准进行修订,以证明将 IPAF 最终指定为独立的诊断术语是合理的,而不是作为临床研究基础提出的暂定实体。这篇综述涵盖了 IPAF 的现状、从 IPAF 证据基础中可以得出和不能得出的结论,以及需要专家组进一步考虑的持续存在的不确定性。