Lacalamita Antonio, Piccinno Emanuele, Scalavino Viviana, Bellotti Roberto, Giannelli Gianluigi, Serino Grazia
National Institute of Gastroenterology "S. de Bellis", Research Hospital, Castellana Grotte, 70013 Bari, Italy.
Dipartimento Interateneo di Fisica, Università degli Studi di Bari Aldo Moro, 70126 Bari, Italy.
Biomedicines. 2021 Dec 17;9(12):1937. doi: 10.3390/biomedicines9121937.
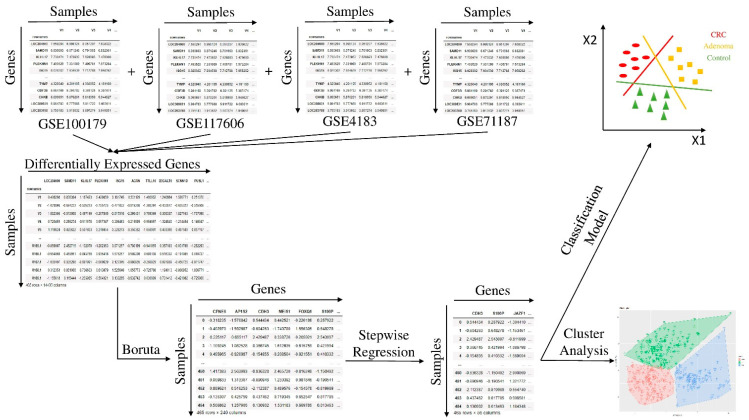
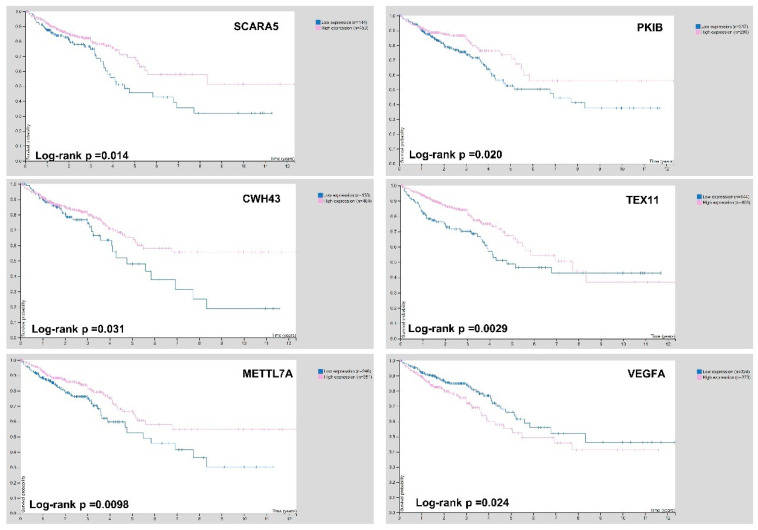
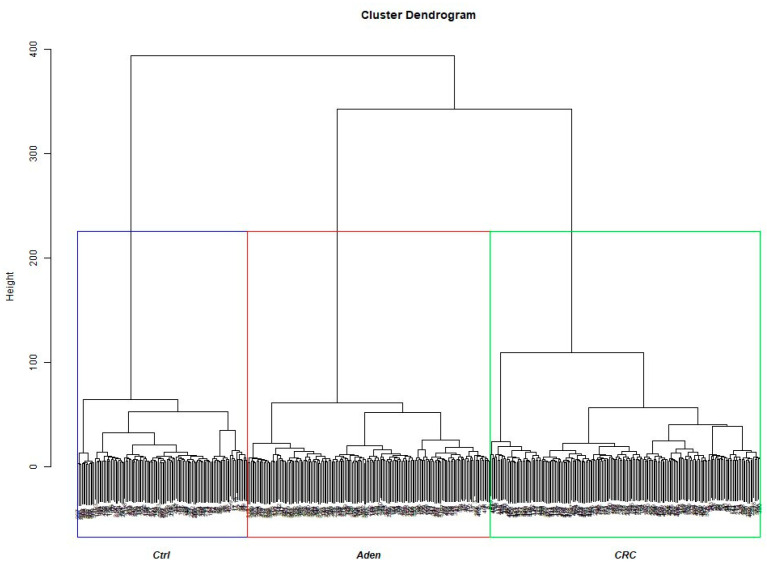
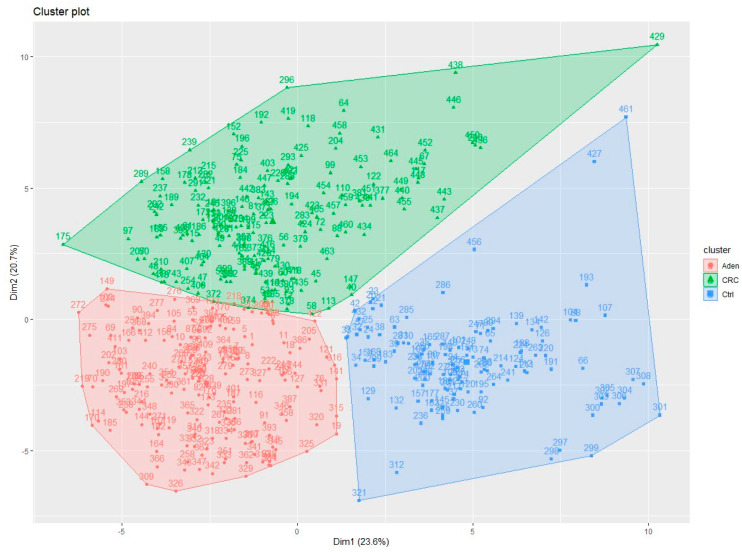
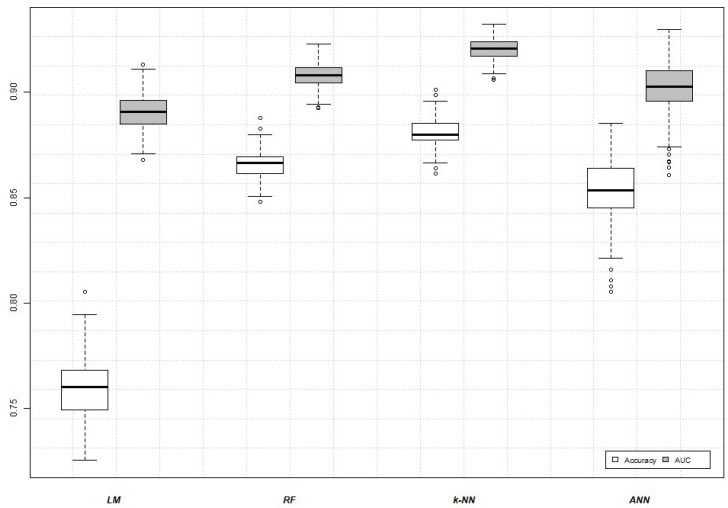
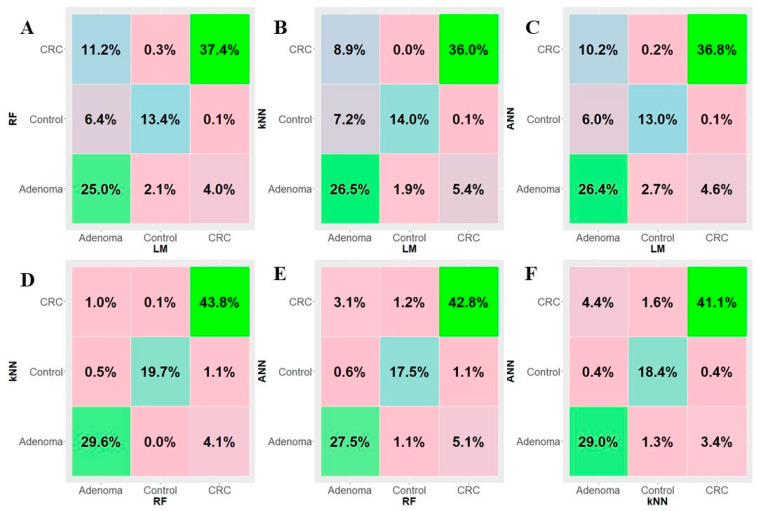
Colorectal cancer (CRC) carcinogenesis is generally the result of the sequential mutation and deletion of various genes; this is known as the normal mucosa-adenoma-carcinoma sequence. The aim of this study was to develop a predictor-classifier during the "adenoma-carcinoma" sequence using microarray gene expression profiles of primary CRC, adenoma, and normal colon epithelial tissues. Four gene expression profiles from the Gene Expression Omnibus database, containing 465 samples (105 normal, 155 adenoma, and 205 CRC), were preprocessed to identify differentially expressed genes (DEGs) between adenoma tissue and primary CRC. The feature selection procedure, using the sequential Boruta algorithm and Stepwise Regression, determined 56 highly important genes. K-Means methods showed that, using the selected 56 DEGs, the three groups were clearly separate. The classification was performed with machine learning algorithms such as Linear Model (LM), Random Forest (RF), k-Nearest Neighbors (k-NN), and Artificial Neural Network (ANN). The best classification method in terms of accuracy (88.06 ± 0.70) and AUC (92.04 ± 0.47) was k-NN. To confirm the relevance of the predictive models, we applied the four models on a validation cohort: the k-NN model remained the best model in terms of performance, with 91.11% accuracy. Among the 56 DEGs, we identified 17 genes with an ascending or descending trend through the normal mucosa-adenoma-carcinoma sequence. Moreover, using the survival information of the TCGA database, we selected six DEGs related to patient prognosis (SCARA5, PKIB, CWH43, TEX11, METTL7A, and VEGFA). The six-gene-based classifier described in the current study could be used as a potential biomarker for the early diagnosis of CRC.
结直肠癌(CRC)的致癌过程通常是多种基因依次发生突变和缺失的结果;这被称为正常黏膜-腺瘤-癌序列。本研究的目的是利用原发性CRC、腺瘤和正常结肠上皮组织的微阵列基因表达谱,在“腺瘤-癌”序列期间开发一种预测分类器。对来自基因表达综合数据库的四个基因表达谱进行了预处理,这些谱包含465个样本(105个正常样本、155个腺瘤样本和205个CRC样本),以识别腺瘤组织和原发性CRC之间的差异表达基因(DEG)。使用顺序博鲁塔算法和逐步回归的特征选择程序确定了56个高度重要的基因。K均值方法表明,使用选定的56个DEG,三组明显分开。使用线性模型(LM)、随机森林(RF)、k近邻(k-NN)和人工神经网络(ANN)等机器学习算法进行分类。就准确率(88.06±0.70)和AUC(92.04±0.47)而言,最佳分类方法是k-NN。为了确认预测模型的相关性,我们在一个验证队列中应用了这四个模型:就性能而言,k-NN模型仍然是最佳模型,准确率为91.11%。在56个DEG中,我们通过正常黏膜-腺瘤-癌序列鉴定出17个呈上升或下降趋势的基因。此外,利用TCGA数据库的生存信息,我们选择了六个与患者预后相关的DEG(SCARA5、PKIB、CWH43、TEX11、METTL7A和VEGFA)。本研究中描述的基于六个基因的分类器可作为CRC早期诊断的潜在生物标志物。