Khayat Michael M, Hu Jianhong, Jiang Yunyun, Li He, Chander Varuna, Dawood Moez, Hansen Adam W, Li Shoudong, Friedman Jennifer, Cross Laura, Bijlsma Emilia K, Ruivenkamp Claudia A L, Sansbury Francis H, Innis Jeffrey W, O'Shea Jessica Omark, Meng Qingchang, Rosenfeld Jill A, McWalter Kirsty, Wangler Michael F, Lupski James R, Posey Jennifer E, Murdock David, Gibbs Richard A
Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX, USA.
Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA.
HGG Adv. 2021 Oct 14;2(4). doi: 10.1016/j.xhgg.2021.100049. Epub 2021 Aug 10.
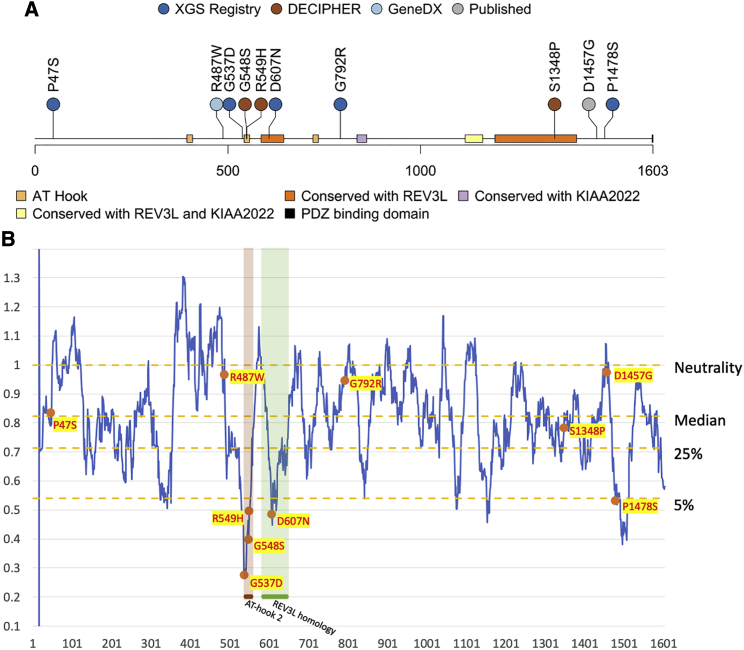
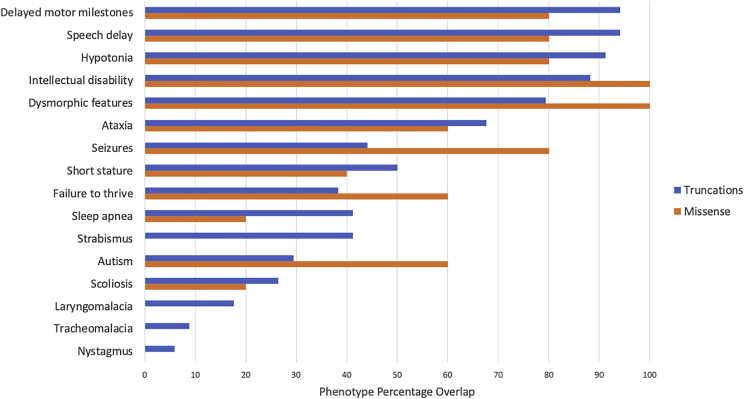
Xia-Gibbs syndrome (XGS; MIM: 615829) is a phenotypically heterogeneous neurodevelopmental disorder (NDD) caused by newly arising mutations in the AT-Hook DNA-Binding Motif-Containing 1 () gene that are predicted to lead to truncated AHDC1 protein synthesis. More than 270 individuals have been diagnosed with XGS worldwide. Despite the absence of an independent assay for AHDC1 protein function to corroborate potential functional consequences of rare variant genetic findings, there are also reports of individuals with XGS-like trait manifestations who have missense mutations and who have been provided a molecular diagnosis of the disorder. To investigate a potential contribution of missense mutations to XGS, we mapped the missense mutations from 10 such individuals to the AHDC1 conserved protein domain structure and detailed the observed phenotypes. Five newly identified individuals were ascertained from a local XGS Registry, and an additional five were taken from external reports or databases, including one publication. Where clinical data were available, individuals with missense mutations all displayed phenotypes consistent with those observed in individuals with truncating mutations, including delayed motor milestones, intellectual disability (ID), hypotonia, and speech delay. A subset of the 10 reported missense mutations cluster in two regions of the AHDC1 protein with known conserved domains, likely representing functional motifs. Variants outside the clustered regions score lower for computational prediction of their likely damaging effects. Overall, missense variants in are likely diagnostic of XGS when analysis of their position relative to conserved regions is considered together with disease trait manifestations.
夏-吉布斯综合征(XGS;MIM:615829)是一种表型异质性神经发育障碍(NDD),由含AT钩DNA结合基序1(AHDC1)基因的新生突变引起,这些突变预计会导致截短的AHDC1蛋白合成。全球已有270多名个体被诊断为XGS。尽管缺乏用于证实罕见变异基因发现潜在功能后果的AHDC1蛋白功能独立检测方法,但也有报道称,具有XGS样性状表现的个体存在错义突变,并已得到该疾病的分子诊断。为了研究错义突变对XGS的潜在影响,我们将10名此类个体的错义突变定位到AHDC1保守蛋白结构域,并详细描述了观察到的表型。从当地的XGS登记处确定了5名新发现的个体,另外5名来自外部报告或数据库,包括一篇出版物。在可获得临床数据的情况下,有错义突变的个体均表现出与截短突变个体一致的表型,包括运动发育迟缓、智力残疾(ID)、肌张力减退和语言发育迟缓。所报告的10个错义突变中的一部分聚集在AHDC1蛋白两个具有已知保守结构域的区域,可能代表功能基序。聚集区域外的变异在计算预测其可能的有害影响时得分较低。总体而言,当结合其相对于保守区域的位置分析以及疾病性状表现时,AHDC1中的错义变异可能有助于XGS的诊断。