Centre for Lipid Research & Key Laboratory of Molecular Biology for Infectious Diseases (Ministry of Education), Institute for Viral Hepatitis, Department of Infectious Diseases, the Second Affiliated Hospital, Chongqing Medical University, 400016 Chongqing, China.
Centre for Lipid Research & Key Laboratory of Molecular Biology for Infectious Diseases (Ministry of Education), Institute for Viral Hepatitis, Department of Infectious Diseases, the Second Affiliated Hospital, Chongqing Medical University, 400016 Chongqing, China; John Moorhead Research Laboratory, Centre for Nephrology, University College London Medical School, Royal Free Campus, University College London, London NW3 2PF, United Kingdom.
Mol Metab. 2022 Mar;57:101428. doi: 10.1016/j.molmet.2021.101428. Epub 2021 Dec 30.
Enhanced de novo lipogenesis (DNL) in hepatocytes is a major contributor to nonalcoholic fatty liver disease (NAFLD). Fatty acid translocase (FAT/CD36) is involved in the pathogenesis of NAFLD through facilitating free fatty acids uptake. Here, we explored the effects of CD36 on DNL and elucidated the underlying mechanisms.
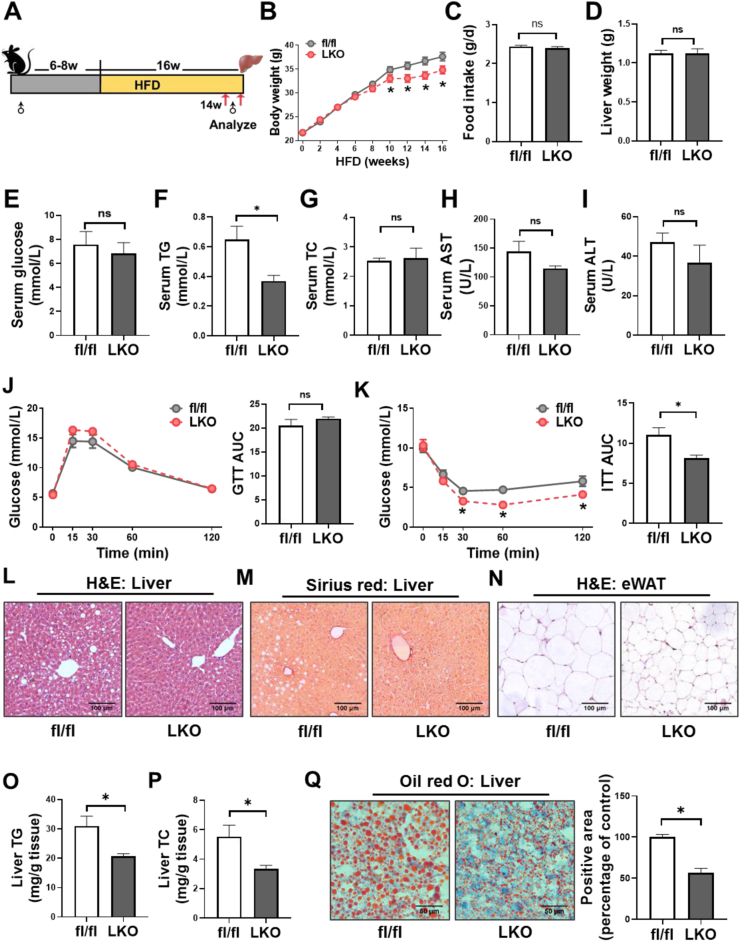
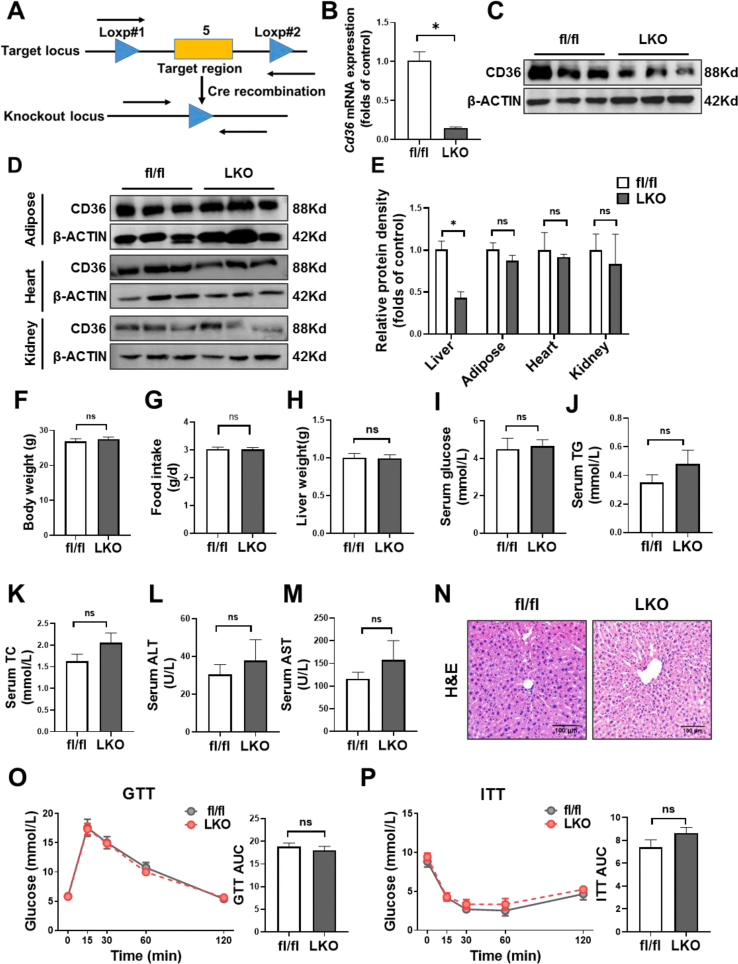
We generated hepatocyte-specific CD36 knockout (CD36LKO) mice to study in vivo effects of CD36 on DNL under high-fat diet (HFD). Lipid deposition and DNL were analyzed in primary hepatocytes isolated from CD36LKO mice or HepG2 cells with CD36 overexpression. RNA sequence, co-immunoprecipitation, and proximity ligation assay were carried out to determine its role in regulating DNL.
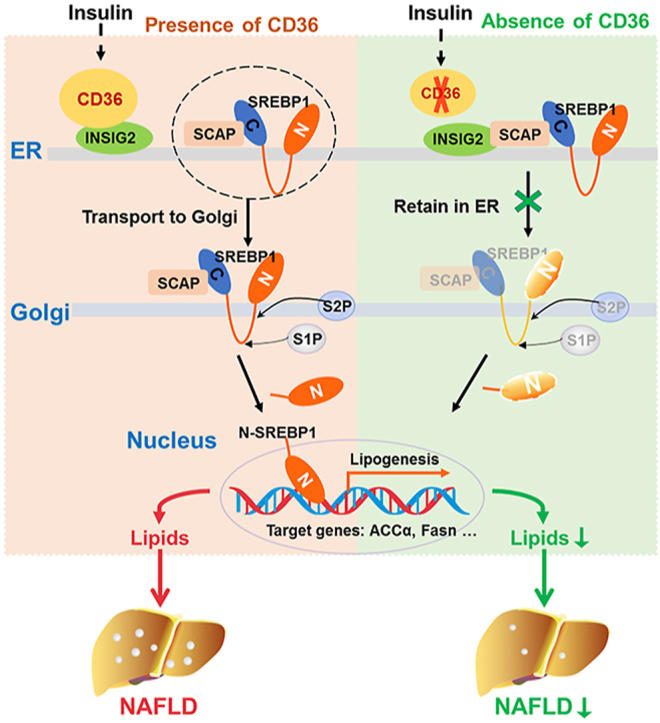
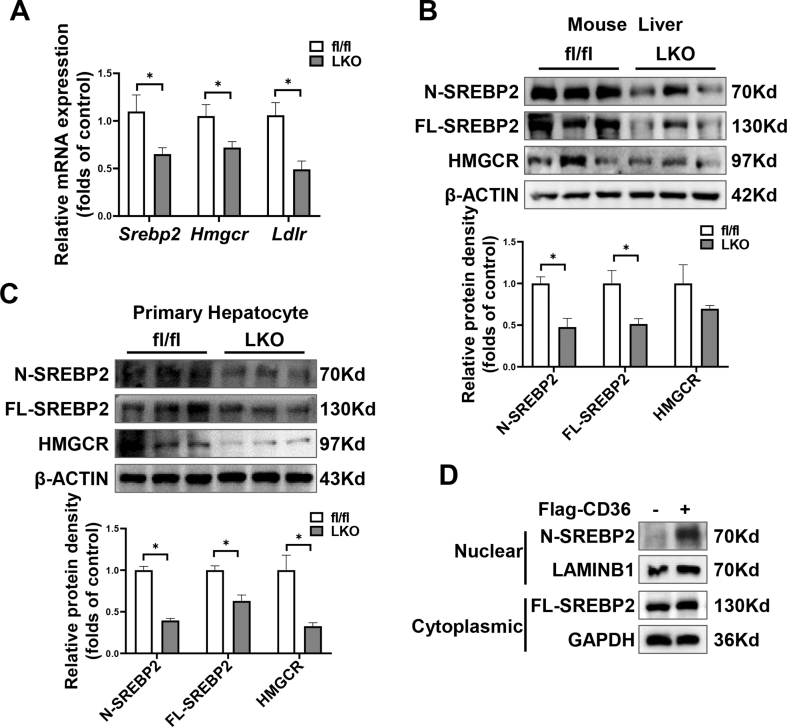
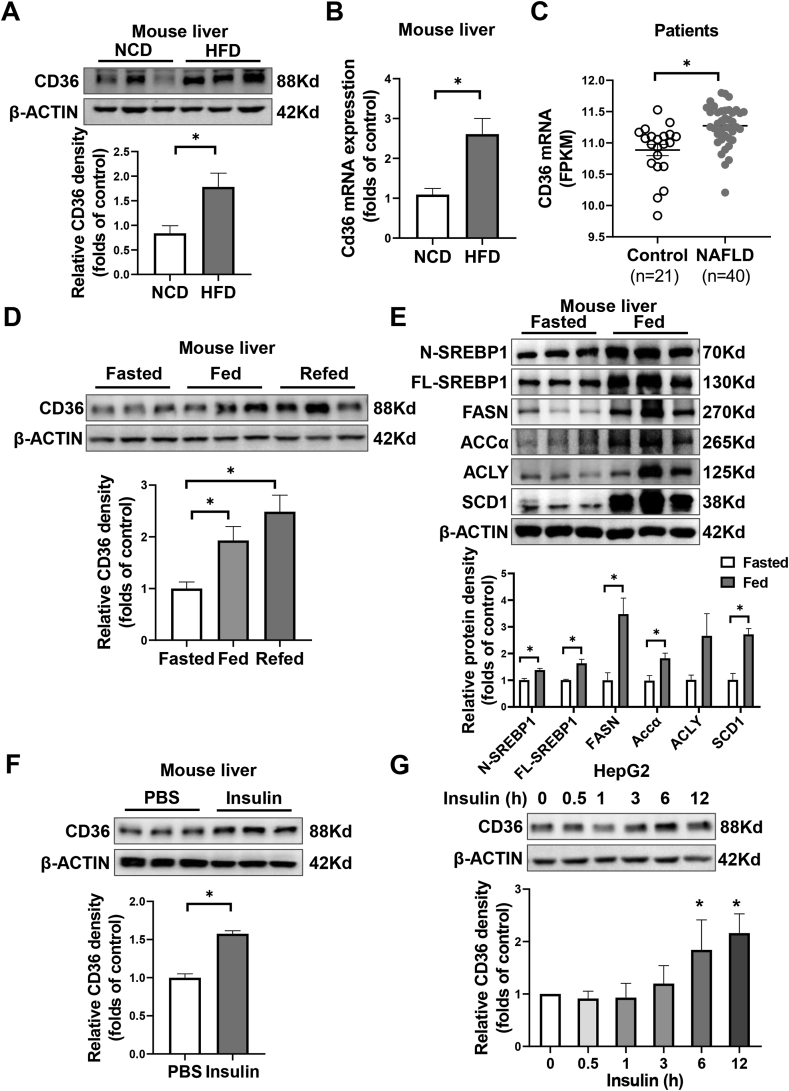
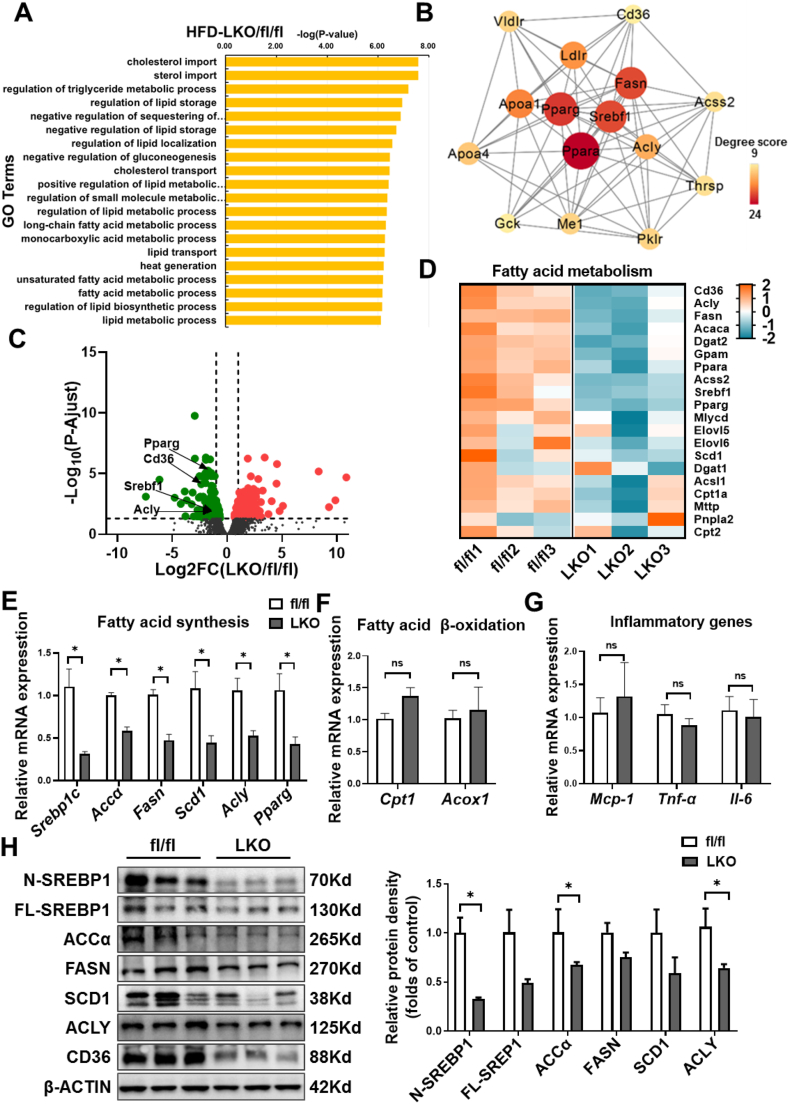
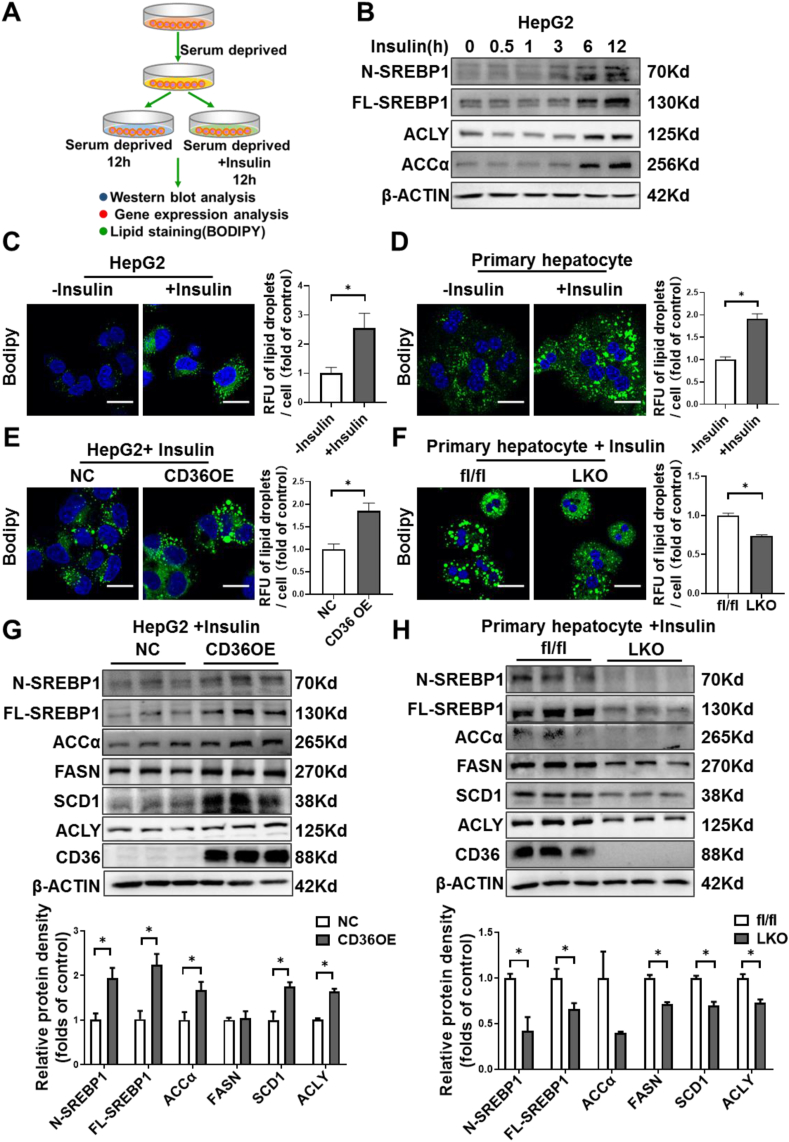
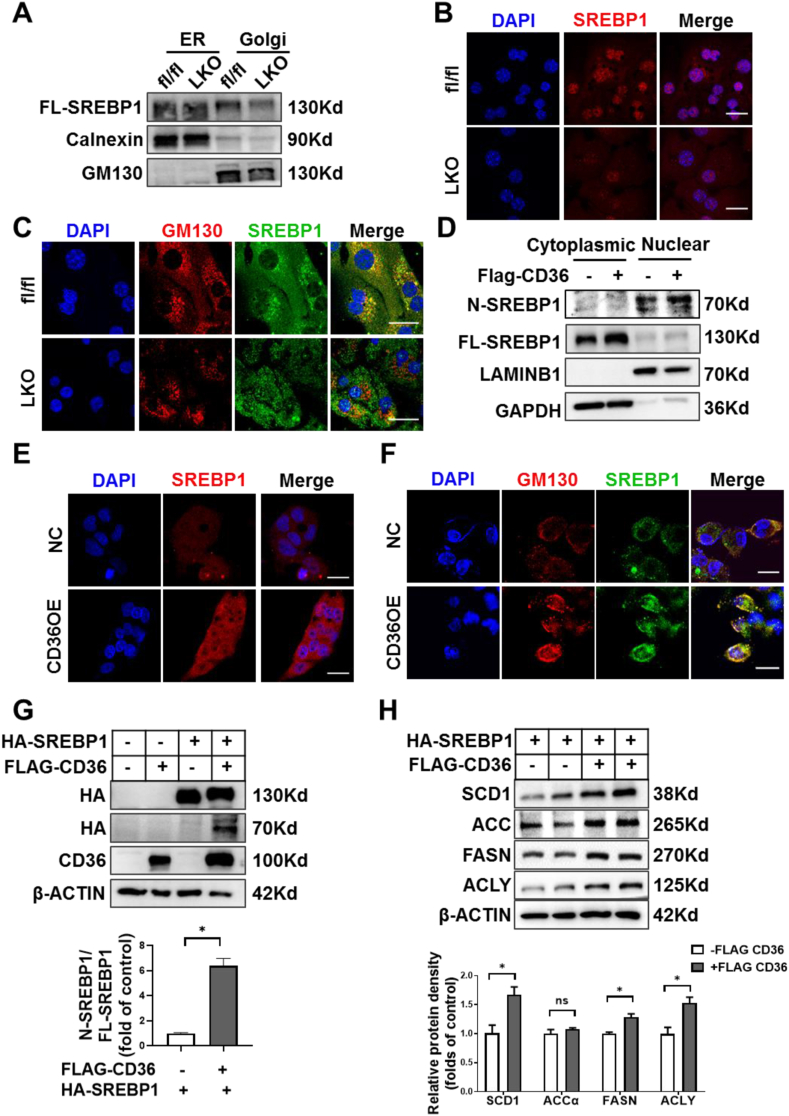
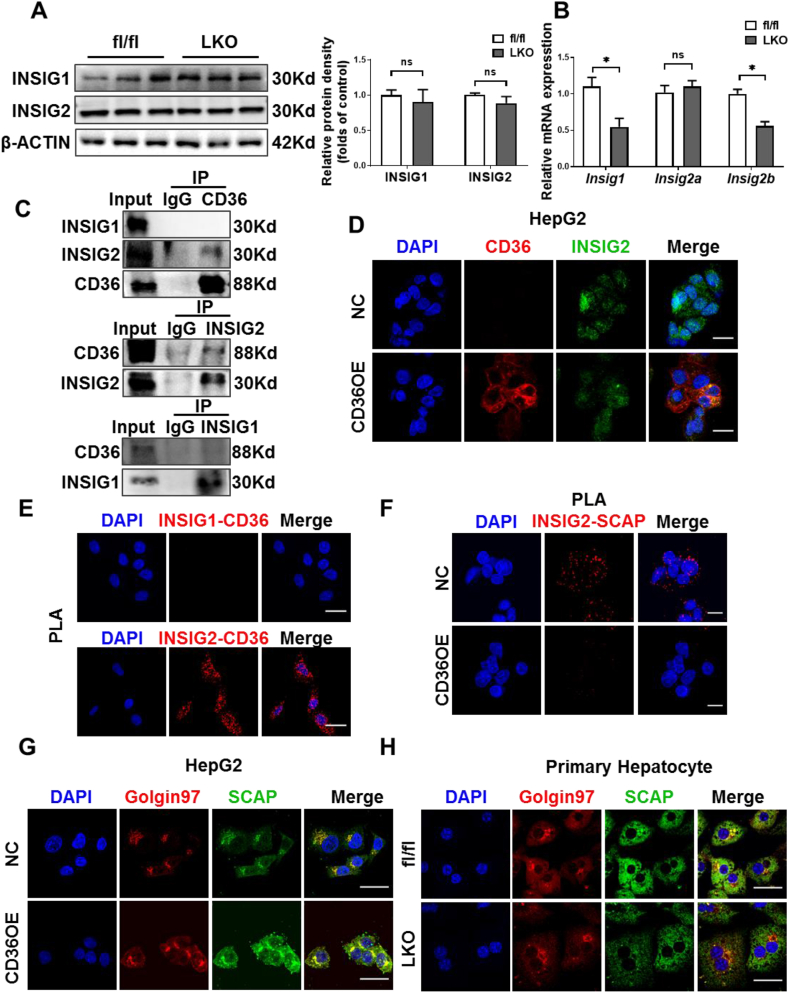
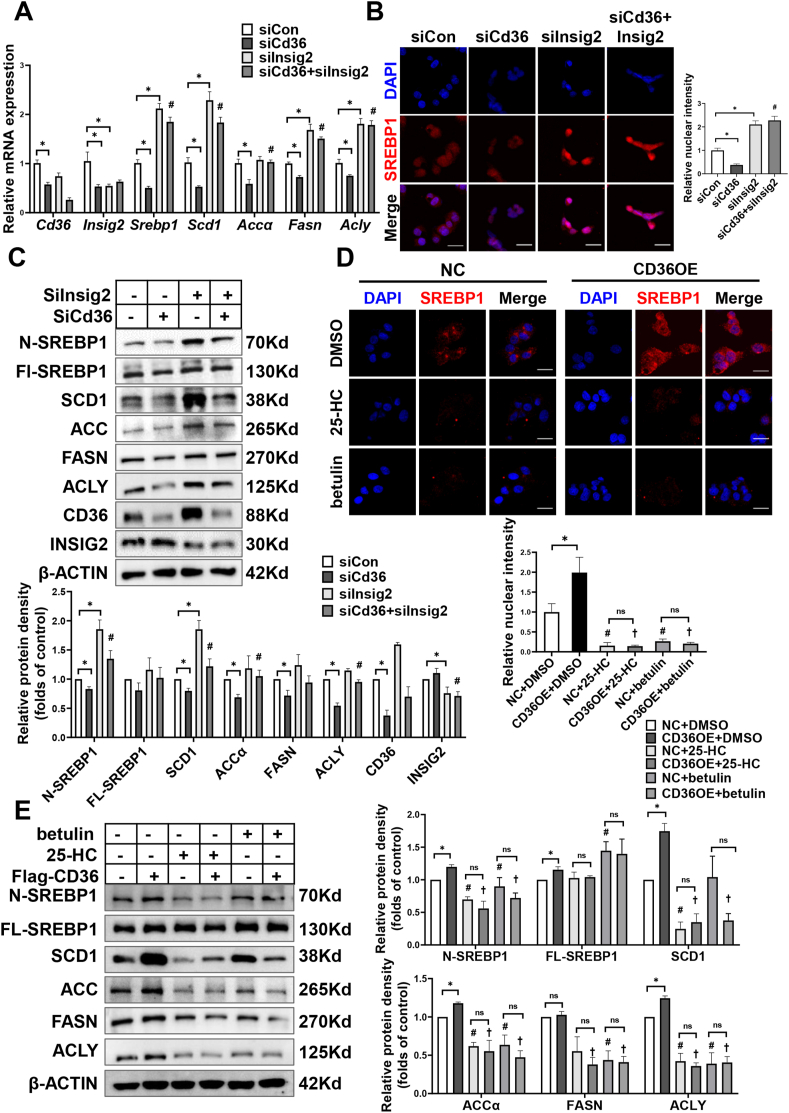
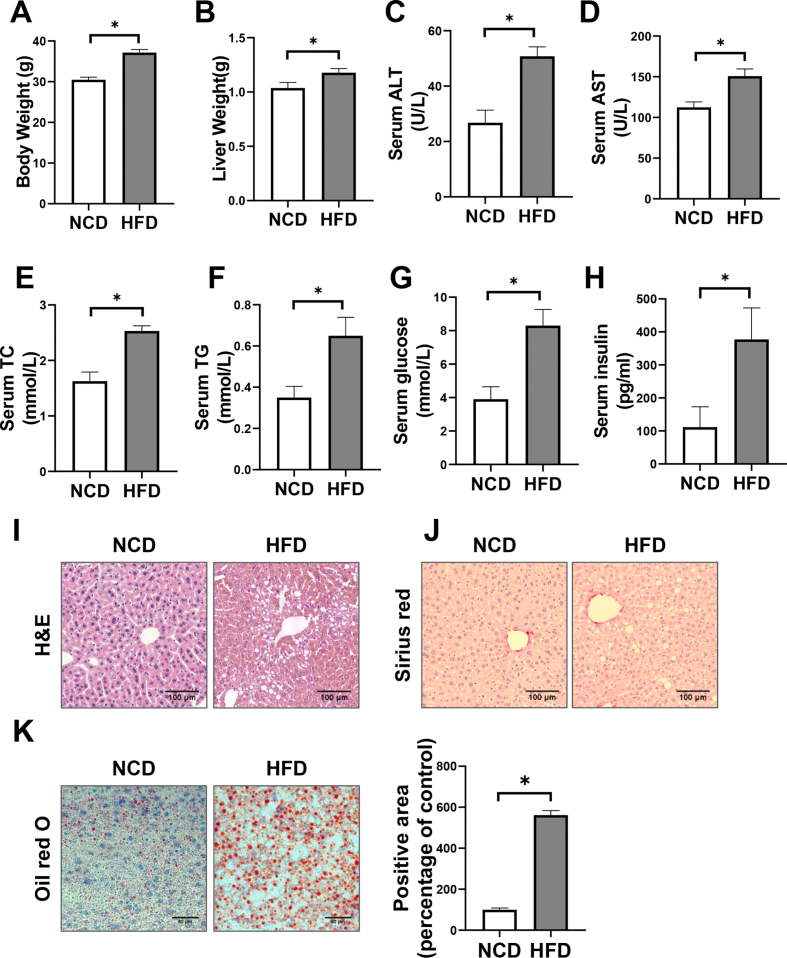
Hepatic CD36 expression was upregulated in NAFLD mice and patients, and CD36LKO mice exhibited attenuated HFD-induced hepatic steatosis and insulin resistance. We identified hepatocyte CD36 as a key regulator for DNL in the liver. Sterol regulatory element-binding protein 1 (SREBP1) and its downstream lipogenic enzymes such as FASN, ACCα, and ACLY were significantly downregulated in the liver of HFD-fed CD36LKO mice, whereas overexpression CD36 stimulated insulin-mediated DNL and lipid droplet formation in vitro. Mechanistically, CD36 was activated by insulin and formed a complex with insulin-induced gene-2 (INSIG2) that disrupts the interaction between SREBP cleavage-activating protein (SCAP) and INSIG2, thereby leading to the translocation of SREBP1 from ER to Golgi for processing. Furthermore, treatment with 25-hydroxycholesterol or betulin molecules shown to enhance SCAP-INSIG interaction, reversed the effects of CD36 on SREBP1 cleavage.
Our findings identify a previously unsuspected role of CD36 in the regulation of hepatic lipogenic program through mediating SREBP1 processing by INSIG2, providing additional evidence for targeting CD36 in NAFLD.
肝细胞中增强的从头脂肪生成(DNL)是导致非酒精性脂肪性肝病(NAFLD)的主要原因。脂肪酸转运蛋白(FAT/CD36)通过促进游离脂肪酸摄取而参与 NAFLD 的发病机制。在这里,我们研究了 CD36 对 DNL 的影响,并阐明了其潜在机制。
我们生成了肝细胞特异性 CD36 敲除(CD36LKO)小鼠,以研究高脂肪饮食(HFD)下 CD36 对 DNL 的体内影响。从 CD36LKO 小鼠或过表达 CD36 的 HepG2 细胞中分离的原代肝细胞中分析脂质沉积和 DNL。进行 RNA 序列、共免疫沉淀和邻近连接分析以确定其在调节 DNL 中的作用。
NAFLD 小鼠和患者的肝 CD36 表达上调,CD36LKO 小鼠表现出 HFD 诱导的肝脂肪变性和胰岛素抵抗减轻。我们确定肝细胞 CD36 是肝脏中 DNL 的关键调节因子。固醇调节元件结合蛋白 1(SREBP1)及其下游脂肪生成酶,如 FASN、ACCα 和 ACLY,在 HFD 喂养的 CD36LKO 小鼠的肝脏中显著下调,而过表达 CD36 可刺激胰岛素介导的 DNL 和体外脂滴形成。在机制上,胰岛素激活 CD36 并与胰岛素诱导基因 2(INSIG2)形成复合物,破坏 SREBP 切割激活蛋白(SCAP)和 INSIG2 之间的相互作用,从而导致 SREBP1 从 ER 易位到高尔基体进行加工。此外,用 25-羟胆固醇或桦木醇处理可增强 SCAP-INSIG 相互作用,逆转 CD36 对 SREBP1 切割的影响。
我们的研究结果确定了 CD36 在调节肝脂肪生成程序中的一个以前未被怀疑的作用,通过 INSIG2 介导 SREBP1 的切割,为 NAFLD 中靶向 CD36 提供了额外的证据。