Department of Pediatrics, Tokyo Metropolitan Kita Medical and Rehabilitation Center for the Disabled, 1-2-3 Jujodai Kita-ku, Tokyo, 114-0033, Japan.
Department of Neurology and Neuropathology (the Brain Bank for Aging Research), Tokyo Metropolitan Geriatric Hospital and Institute of Gerontology, Tokyo, Japan, 35-2 Sakae-cho, Itabashi-ku, Tokyo, 173-0015, Japan.
BMC Neurol. 2022 Jan 3;22(1):2. doi: 10.1186/s12883-021-02514-z.
The detailed neuropathological features of patients with autosomal recessive hereditary spastic paraplegia with a thin corpus callosum (TCC) and SPG11 mutations are poorly understood, as only a few autopsies have been reported. Herein, we describe the clinicopathological findings of a patient with this disease who received long-term care at our medical facility.
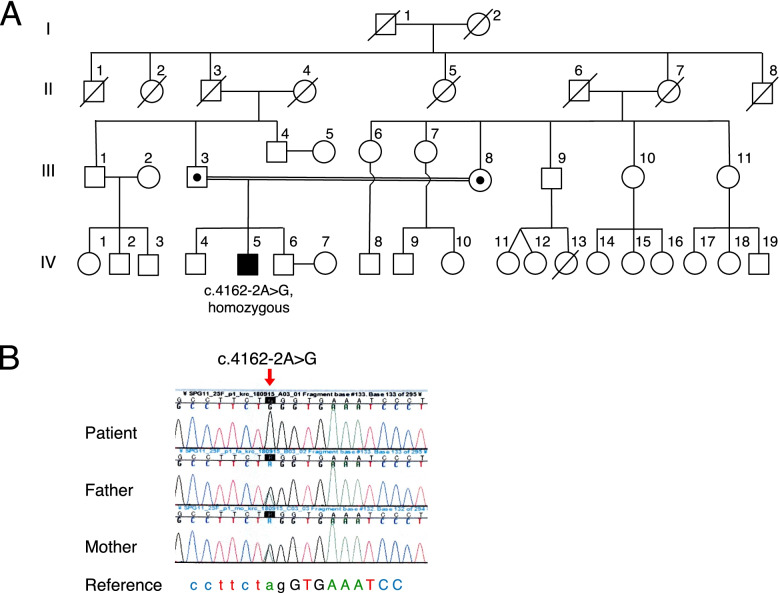
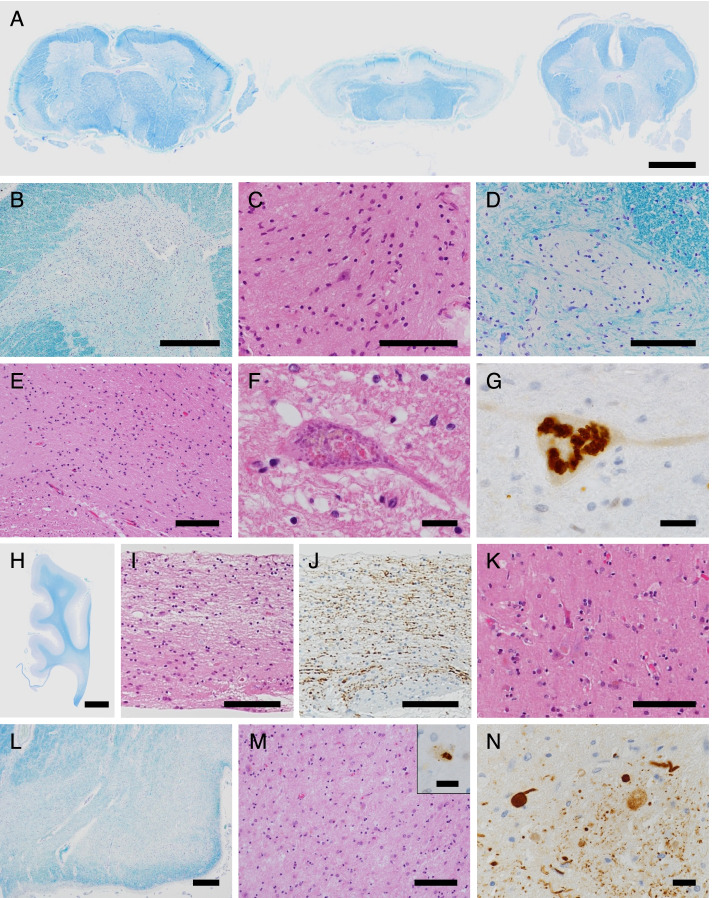
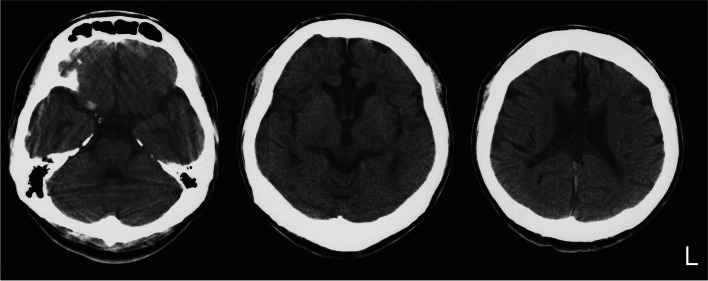
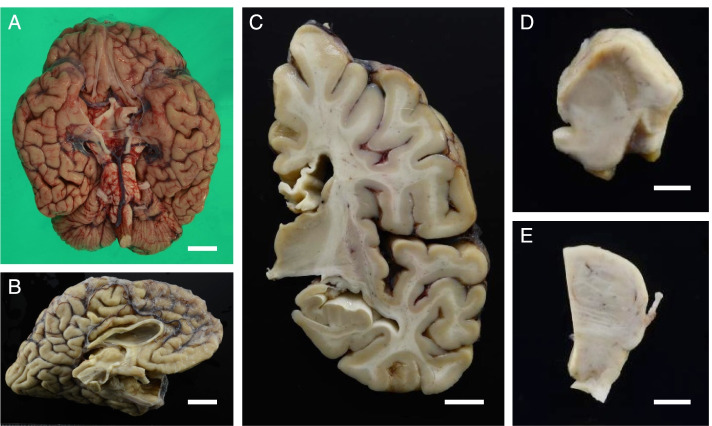
A Japanese man exhibited a mild developmental delay in early childhood and intellectual disability, followed by the appearance of a spastic gait by age 13. At the age of 25 years, he became bedridden and needed a ventilator. Genetic analysis revealed a homozygous splice site variant in the SPG11 gene (c. 4162-2A > G) after the provision of genetic counselling and acquisition of informed consent from his parents. He died of pneumonia at the age of 44. His brain weighed 967 g and was characterized by a TCC, and his spinal cord was flattened. Microscopically, degeneration was observed in the posterior spinocerebellar tract, the gracile fasciculus, and the posterior column in addition to the corticospinal tract. Marked neuronal loss and gliosis were observed in the anterior horn, Clarke's column, and hypoglossal and facial nuclei. Various types of neurons, in addition to motor neurons, showed coarse eosinophilic granules that were immunoreactive for p62. The loss of pigmented neurons with gliosis was apparent in both the substantia nigra and locus coeruleus. Lateral geniculate body degeneration was a characteristic feature of this patient. Furthermore, peripheral Lewy body-related α-synucleinopathy and scattered α-synuclein-immunoreactive neurites in the locus coeruleus and reticular formation of the brainstem were observed.
In patients with hereditary spastic paraplegia with SPG11 mutations, a variety of clinical phenotypes develop due to widespread lesions containing p62-immunoreactive neuronal cytoplasmic inclusions. We herein report the lateral geniculate body as another degenerative site related to SPG11-related pathologies that should be studied in future investigations.
携带 SPG11 突变的常染色体隐性遗传性痉挛性截瘫伴薄胼胝体(TCC)患者的详细神经病理学特征了解甚少,因为仅有少数尸检报道过此类病例。在此,我们描述了一位在我们医疗机构接受长期护理的该病患者的临床病理发现。
一名日本男性在幼儿期表现出轻度发育迟缓及智力障碍,13 岁时出现痉挛性步态。25 岁时,他卧床不起,需要使用呼吸机。在提供遗传咨询并获得其父母的知情同意后,基因分析发现 SPG11 基因的纯合剪接位点变异(c.4162-2A > G)。他 44 岁时因肺炎去世。其大脑重 967g,表现为 TCC,脊髓变平。显微镜下,除皮质脊髓束外,后索小脑束、薄束和后柱也有变性。前角、Clarke 柱、舌下核和面神经核可见明显神经元丢失和神经胶质增生。除运动神经元外,各种类型的神经元均可见粗大嗜酸性颗粒,p62 免疫反应阳性。黑质和蓝斑内色素性神经元丢失伴神经胶质增生明显。外侧膝状体变性是该患者的一个特征性表现。此外,还观察到脑桥蓝斑和网状结构存在与外周路易体相关的α-突触核蛋白病和散在的α-突触核蛋白免疫反应性神经原纤维。
携带 SPG11 突变的遗传性痉挛性截瘫患者由于存在包含 p62 免疫反应性神经元胞质内含物的广泛病变,可出现多种临床表现。我们在此报告外侧膝状体是与 SPG11 相关病理学相关的另一个变性部位,未来的研究应进一步探讨。