Denora Paola S, Smets Katrien, Zolfanelli Federica, Ceuterick-de Groote Chantal, Casali Carlo, Deconinck Tine, Sieben Anne, Gonzales Michael, Zuchner Stephan, Darios Frédéric, Peeters Dirk, Brice Alexis, Malandrini Alessandro, De Jonghe Peter, Santorelli Filippo M, Stevanin Giovanni, Martin Jean-Jacques, El Hachimi Khalid H
1 Ecole Pratique des Hautes Etudes, EPHE, PSL université, laboratoire de neurogénétique, F-75013, Paris, France 2 Inserm, U1127, F-75013, Paris, France 3 CNRS, UMR7225, F-75013, Paris, France 4 Sorbonne Universités, UPMC Univ Paris 06, UMR_S1127, Institut du Cerveau et de la Moelle épinière - ICM, Pitié-Salpêtrière Hospital, F-75013, Paris, France 5 Department of Genetics and Rare Diseases, IRCCS Bambino Gesu' Children Hospital, Rome, Italy.
6 Neurogenetics Group, VIB-Department of Molecular Genetics, University of Antwerp, Belgium 7 Laboratories of Neurogenetics, Institute Born-Bunge, University of Antwerp, Belgium 8 Department of Neurology, Antwerp University Hospital, Antwerp, Belgium.
Brain. 2016 Jun;139(Pt 6):1723-34. doi: 10.1093/brain/aww061. Epub 2016 Mar 25.
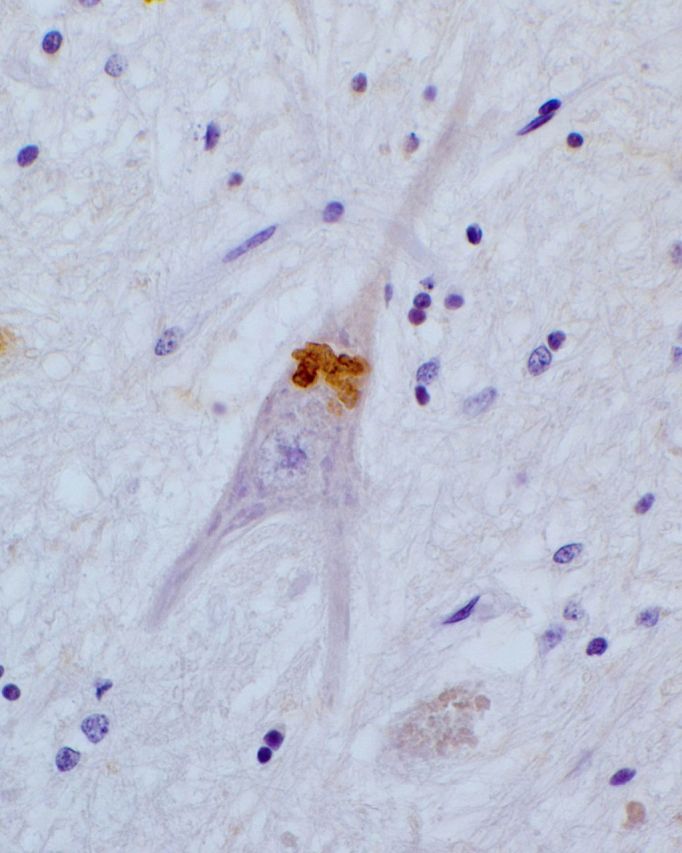
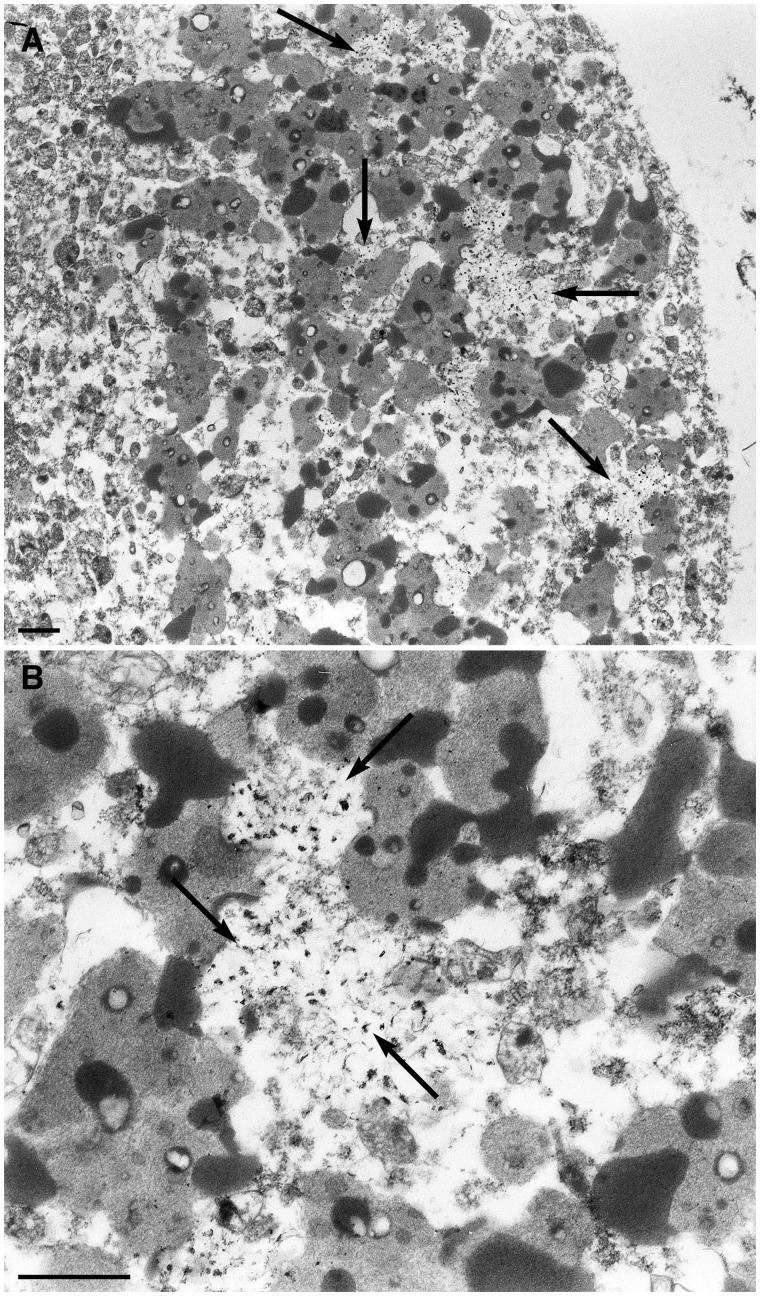
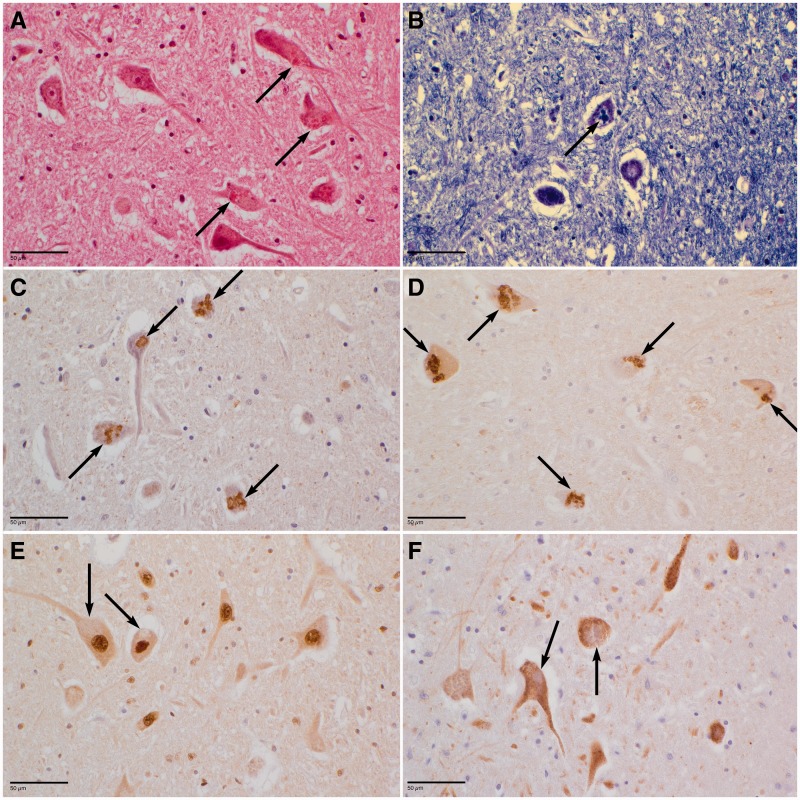
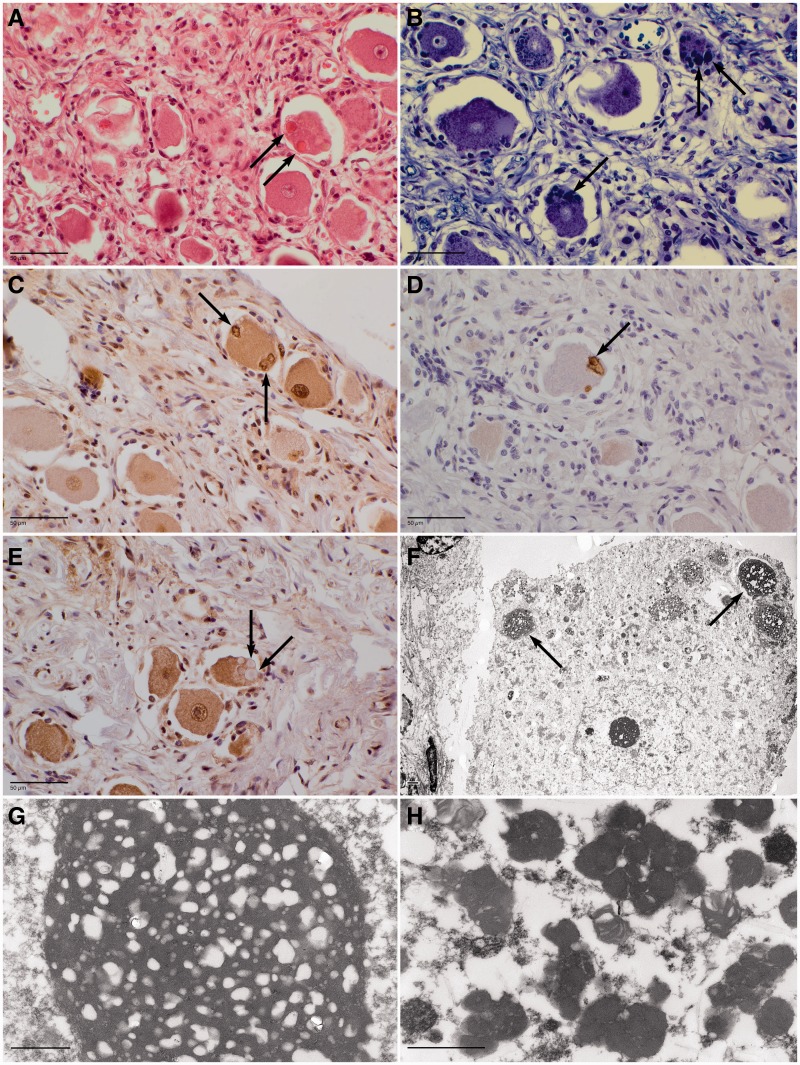
The most common form of autosomal recessive hereditary spastic paraplegia is caused by mutations in the SPG11/KIAA1840 gene on chromosome 15q. The nature of the vast majority of SPG11 mutations found to date suggests a loss-of-function mechanism of the encoded protein, spatacsin. The SPG11 phenotype is, in most cases, characterized by a progressive spasticity with neuropathy, cognitive impairment and a thin corpus callosum on brain MRI. Full neuropathological characterization has not been reported to date despite the description of >100 SPG11 mutations. We describe here the clinical and pathological features observed in two unrelated females, members of genetically ascertained SPG11 families originating from Belgium and Italy, respectively. We confirm the presence of lesions of motor tracts in medulla oblongata and spinal cord associated with other lesions of the central nervous system. Interestingly, we report for the first time pathological hallmarks of SPG11 in neurons that include intracytoplasmic granular lysosome-like structures mainly in supratentorial areas, and others in subtentorial areas that are partially reminiscent of those observed in amyotrophic lateral sclerosis, such as ubiquitin and p62 aggregates, except that they are never labelled with anti-TDP-43 or anti-cystatin C. The neuropathological overlap with amyotrophic lateral sclerosis, associated with some shared clinical manifestations, opens up new fields of investigation in the physiopathological continuum of motor neuron degeneration.
常染色体隐性遗传性痉挛性截瘫最常见的形式是由15号染色体q臂上的SPG11/KIAA1840基因突变引起的。迄今为止发现的绝大多数SPG11突变的性质表明,其编码蛋白spatacsin存在功能丧失机制。在大多数情况下,SPG11的表型特征为进行性痉挛伴神经病变、认知障碍以及脑部MRI显示胼胝体变薄。尽管已描述了100多种SPG11突变,但迄今为止尚未有完整的神经病理学特征报告。我们在此描述了分别来自比利时和意大利的两个经基因确定的SPG11家族中两名无关女性的临床和病理特征。我们证实了延髓和脊髓中运动束病变与中枢神经系统的其他病变相关。有趣的是,我们首次报告了SPG11在神经元中的病理特征,包括主要位于幕上区域的胞浆内颗粒状溶酶体样结构,以及幕下区域的其他结构,这些结构部分让人联想到肌萎缩侧索硬化中观察到的结构,如泛素和p62聚集体,但它们从未被抗TDP-43或抗胱抑素C标记。与肌萎缩侧索硬化的神经病理学重叠,以及一些共同的临床表现,为运动神经元变性的生理病理连续体开辟了新的研究领域。