Center for Virology, Medical University of Vienna, Vienna, Austria.
Groningen Institute for Evolutionary Life Sciences, University of Groningen, Groningen, Netherlands.
BMC Genomics. 2022 Jan 6;23(1):31. doi: 10.1186/s12864-021-08272-z.
Short read sequencing has been used extensively to decipher the genome diversity of human cytomegalovirus (HCMV) strains, but falls short to reveal individual genomes in mixed HCMV strain populations. Novel third-generation sequencing platforms offer an extended read length and promise to resolve how distant polymorphic sites along individual genomes are linked. In the present study, we established a long amplicon PacBio sequencing workflow to identify the absolute and relative quantities of unique HCMV haplotypes spanning over multiple hypervariable sites in mixtures. Initial validation of this approach was performed with defined HCMV DNA templates derived from cell-culture enriched viruses and was further tested for its suitability on patient samples carrying mixed HCMV infections.
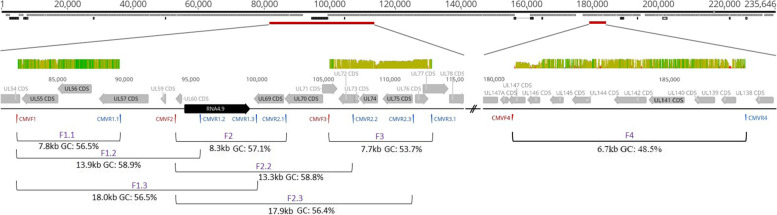
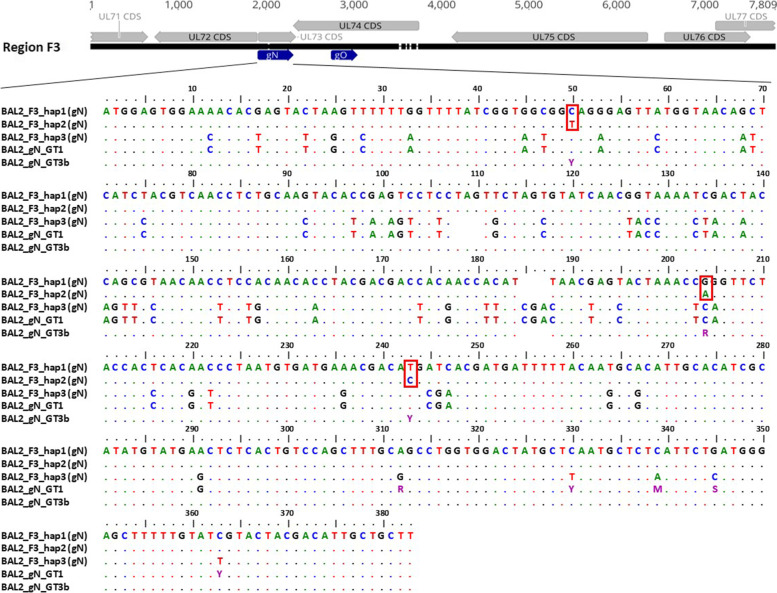
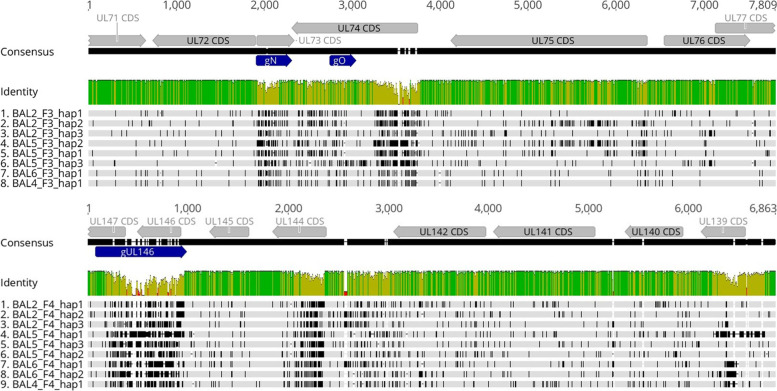
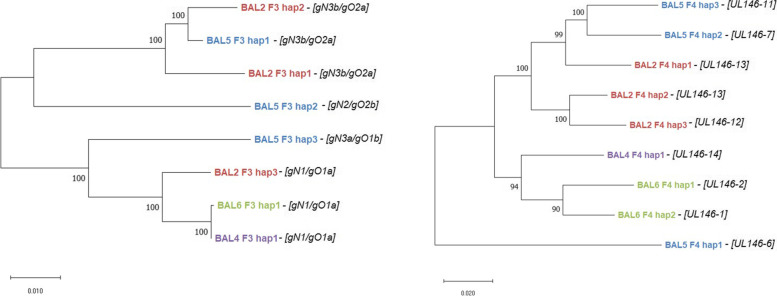
Total substitution and indel error rate of mapped reads ranged from 0.17 to 0.43% depending on the stringency of quality trimming. Artificial HCMV DNA mixtures were correctly determined down to 1% abundance of the minor DNA source when the total HCMV DNA input was 4 × 10 copies/ml. PCR products of up to 7.7 kb and a GC content < 55% were efficiently generated when DNA was directly isolated from patient samples. In a single sample, up to three distinct haplotypes were identified showing varying relative frequencies. Alignments of distinct haplotype sequences within patient samples showed uneven distribution of sequence diversity, interspersed by long identical stretches. Moreover, diversity estimation at single polymorphic regions as assessed by short amplicon sequencing may markedly underestimate the overall diversity of mixed haplotype populations.
Quantitative haplotype determination by long amplicon sequencing provides a novel approach for HCMV strain characterisation in mixed infected samples which can be scaled up to cover the majority of the genome by multi-amplicon panels. This will substantially improve our understanding of intra-host HCMV strain diversity and its dynamic behaviour.
短读测序技术已广泛用于破译人类巨细胞病毒(HCMV)株的基因组多样性,但无法揭示混合 HCMV 株群体中的个体基因组。新型第三代测序平台提供了更长的读长,并有望解决个体基因组中沿多个高度多态性位点的遥远多态性位点如何相关联的问题。在本研究中,我们建立了长扩增子 PacBio 测序工作流程,以鉴定跨越多个高变位点的混合 HCMV 中独特 HCMV 单倍型的绝对和相对数量。该方法的初步验证是使用源自细胞培养富集病毒的明确 HCMV DNA 模板进行的,并进一步测试了其在携带混合 HCMV 感染的患者样本中的适用性。
映射读的总替换和插入缺失错误率取决于质量修剪的严格程度,范围为 0.17%至 0.43%。当总 HCMV DNA 输入为 4×10 拷贝/ml 时,人工 HCMV DNA 混合物可正确确定低至 1%丰度的次要 DNA 源。当直接从患者样本中提取 DNA 时,可有效生成长达 7.7 kb 的 PCR 产物,且 GC 含量<55%。在单个样本中,可鉴定多达三种不同的单倍型,显示出不同的相对频率。患者样本中单倍型序列的比对显示序列多样性分布不均匀,其间散布着长的相同序列。此外,通过短扩增子测序评估的单个多态性区域的多样性估计可能会大大低估混合单倍型群体的总体多样性。
长扩增子测序的定量单倍型测定为混合感染样本中 HCMV 株特征提供了一种新方法,该方法可以通过多扩增子面板扩展到覆盖基因组的大部分,从而大大提高我们对宿主内 HCMV 株多样性及其动态行为的理解。