Peter MacCallum Cancer Centre, Victoria, Australia.
School of BioSciences, University of Melbourne, Victoria, Australia.
Genome Biol. 2022 Jan 6;23(1):10. doi: 10.1186/s13059-021-02588-5.
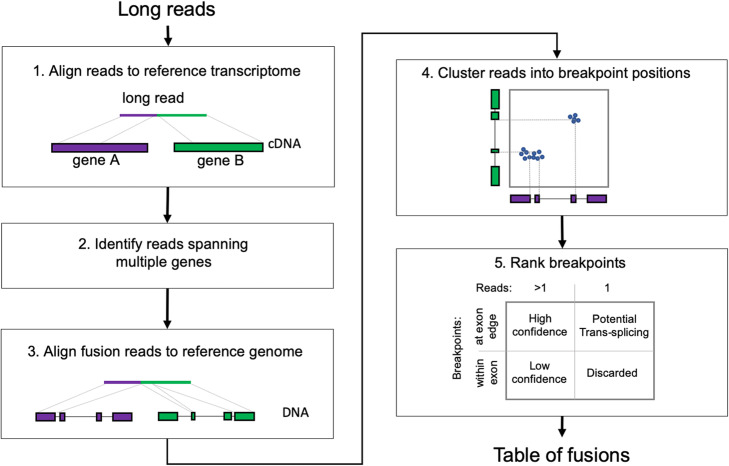
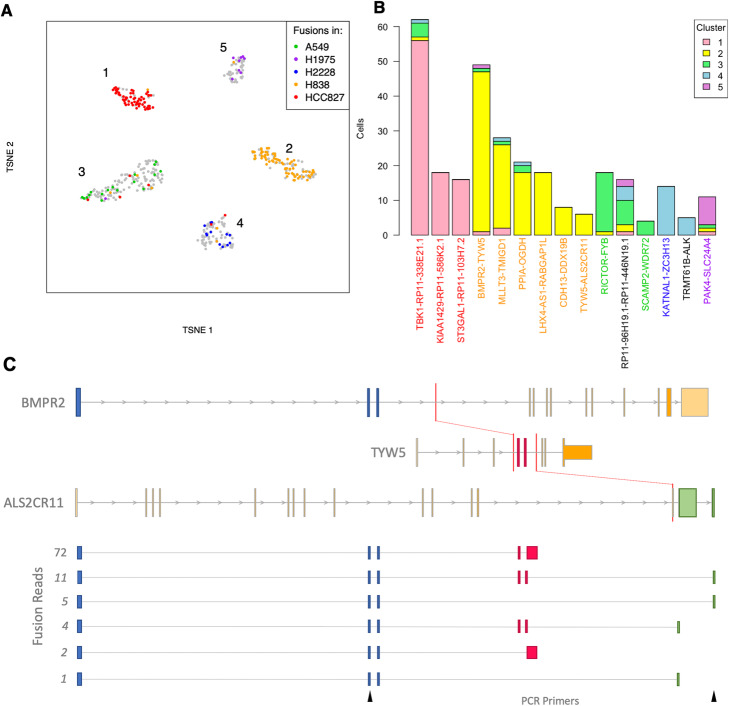
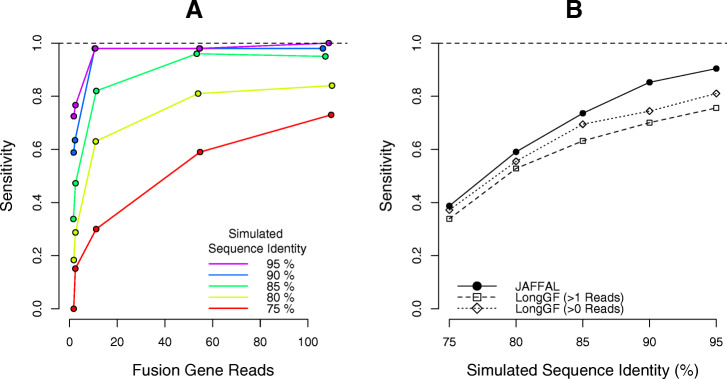
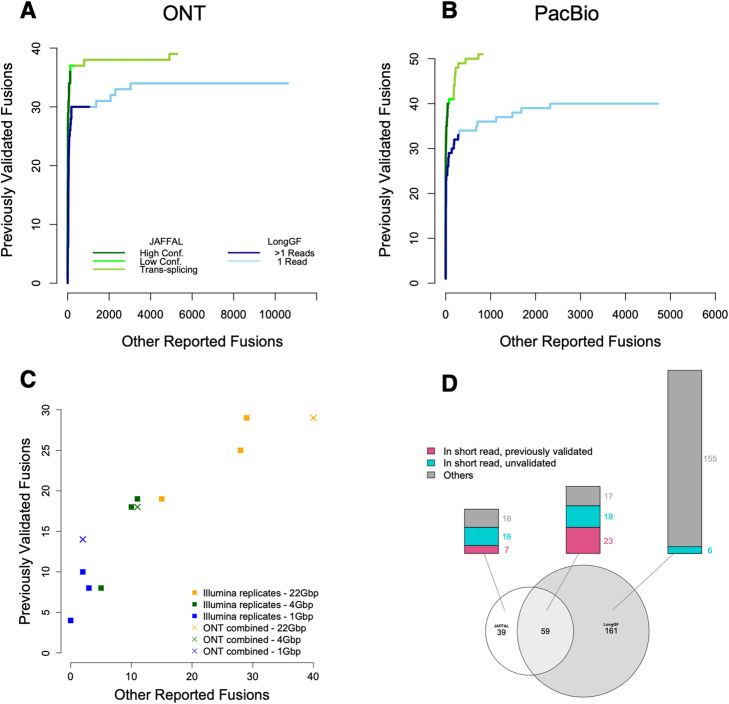
In cancer, fusions are important diagnostic markers and targets for therapy. Long-read transcriptome sequencing allows the discovery of fusions with their full-length isoform structure. However, due to higher sequencing error rates, fusion finding algorithms designed for short reads do not work. Here we present JAFFAL, to identify fusions from long-read transcriptome sequencing. We validate JAFFAL using simulations, cell lines, and patient data from Nanopore and PacBio. We apply JAFFAL to single-cell data and find fusions spanning three genes demonstrating transcripts detected from complex rearrangements. JAFFAL is available at https://github.com/Oshlack/JAFFA/wiki .
在癌症中,融合是重要的诊断标志物和治疗靶点。长读长转录组测序允许发现具有全长异构体结构的融合。然而,由于测序错误率较高,专为短读长设计的融合发现算法无法正常工作。本文中,我们提出了 JAFFAL,用于从长读长转录组测序中识别融合。我们使用模拟数据、细胞系和 Nanopore 和 PacBio 的患者数据对 JAFFAL 进行了验证。我们将 JAFFAL 应用于单细胞数据,并发现了跨越三个基因的融合,这些融合展示了从复杂重排中检测到的转录本。JAFFAL 可在 https://github.com/Oshlack/JAFFA/wiki 上获取。