Vellichirammal Neetha Nanoth, Albahrani Abrar, Banwait Jasjit K, Mishra Nitish K, Li You, Roychoudhury Shrabasti, Kling Mathew J, Mirza Sameer, Bhakat Kishor K, Band Vimla, Joshi Shantaram S, Guda Chittibabu
Department of Genetics, Cell Biology and Anatomy, University of Nebraska Medical Center, Omaha, NE 68198, USA.
Department of Genetics, Cell Biology and Anatomy, University of Nebraska Medical Center, Omaha, NE 68198, USA.
Mol Ther Nucleic Acids. 2020 Mar 6;19:1379-1398. doi: 10.1016/j.omtn.2020.01.023. Epub 2020 Jan 29.
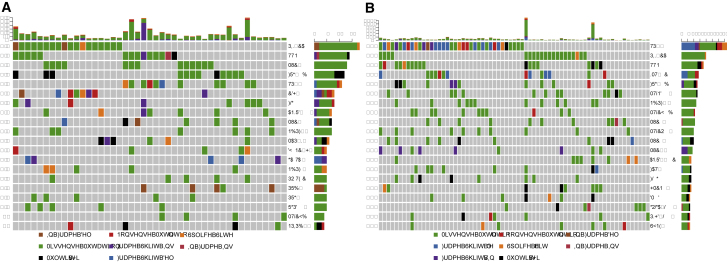
Gene fusions that contribute to oncogenicity can be explored for identifying cancer biomarkers and potential drug targets. To investigate the nature and distribution of fusion transcripts in cancer, we examined the transcriptome data of about 9,000 primary tumors from 33 different cancers in TCGA (The Cancer Genome Atlas) along with cell line data from CCLE (Cancer Cell Line Encyclopedia) using ChimeRScope, a novel fusion detection algorithm. We identified several fusions with sense (canonical, 39%) or antisense (non-canonical, 61%) transcripts recurrent across cancers. The majority of the recurrent non-canonical fusions found in our study are novel, unexplored, and exhibited highly variable profiles across cancers, with breast cancer and glioblastoma having the highest and lowest rates, respectively. Overall, 4,344 recurrent fusions were identified from TCGA in this study, of which 70% were novel. Additional analysis of 802 tumor-derived cell line transcriptome data across 20 cancers revealed significant variability in recurrent fusion profiles between primary tumors and corresponding cell lines. A subset of canonical and non-canonical fusions was validated by examining the structural variation evidence in whole-genome sequencing (WGS) data or by Sanger sequencing of fusion junctions. Several recurrent fusion genes identified in our study show promise for drug repurposing in basket trials and present opportunities for mechanistic studies.
有助于致癌性的基因融合可用于识别癌症生物标志物和潜在药物靶点。为了研究癌症中融合转录本的性质和分布,我们使用一种新型融合检测算法ChimeRScope,检查了来自TCGA(癌症基因组图谱)中33种不同癌症的约9000个原发性肿瘤的转录组数据以及来自CCLE(癌细胞系百科全书)的细胞系数据。我们鉴定出了几种在癌症中反复出现的具有正义(典型,39%)或反义(非典型,61%)转录本的融合。我们研究中发现的大多数反复出现的非典型融合是新颖的、未被探索的,并且在不同癌症中表现出高度可变的特征,乳腺癌和胶质母细胞瘤的发生率分别最高和最低。总体而言,本研究从TCGA中鉴定出4344个反复出现的融合,其中70%是新颖的。对20种癌症的802个肿瘤来源细胞系转录组数据的进一步分析揭示了原发性肿瘤和相应细胞系之间反复出现的融合特征存在显著差异。通过检查全基因组测序(WGS)数据中的结构变异证据或通过融合接头的桑格测序,对一部分典型和非典型融合进行了验证。我们研究中鉴定出的几种反复出现的融合基因在篮子试验中显示出药物再利用的前景,并为机制研究提供了机会。