Public Health Sciences Division, Fred Hutchinson Cancer Research Center, Seattle, WA, USA.
Department of Epidemiology, University of Washington, Seattle, WA, USA.
Sci Rep. 2022 Jan 7;12(1):127. doi: 10.1038/s41598-021-03945-x.
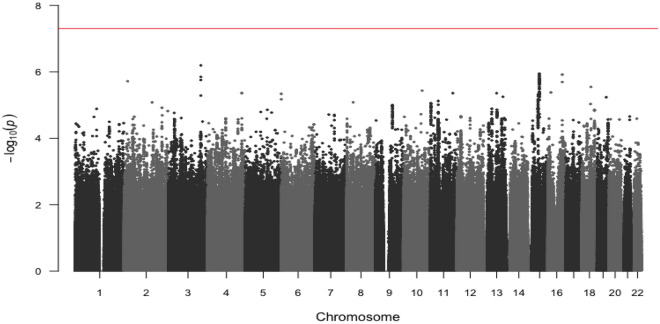
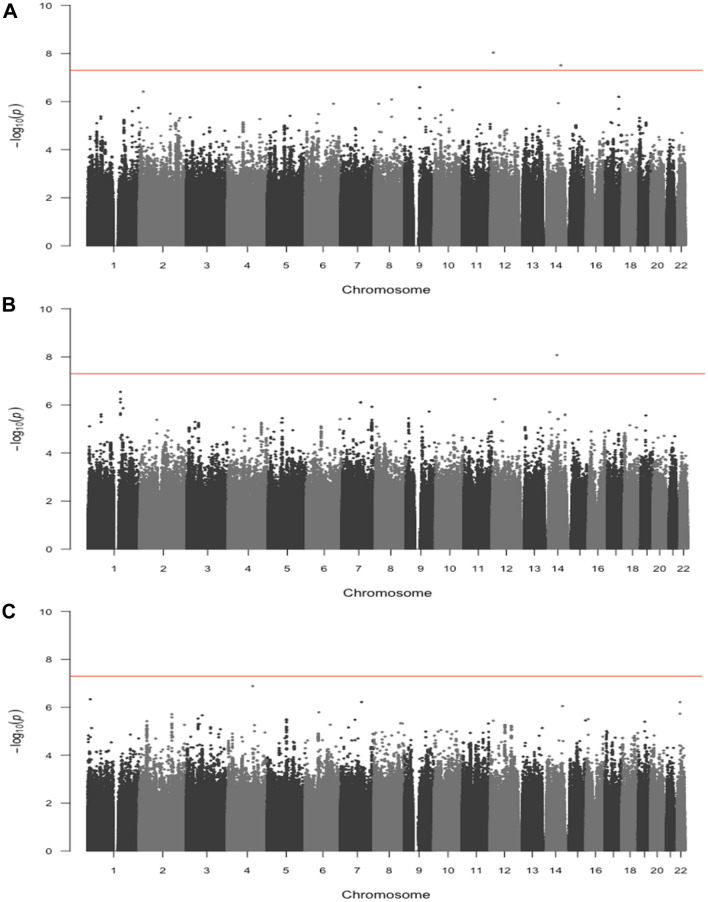
Identification of new genetic markers may improve the prediction of colorectal cancer prognosis. Our objective was to examine genome-wide associations of germline genetic variants with disease-specific survival in an analysis of 16,964 cases of colorectal cancer. We analyzed genotype and colorectal cancer-specific survival data from a consortium of 15 studies. Approximately 7.5 million SNPs were examined under the log-additive model using Cox proportional hazards models, adjusting for clinical factors and principal components. Additionally, we ran secondary analyses stratifying by tumor site and disease stage. We used a genome-wide p-value threshold of 5 × 10 to assess statistical significance. No variants were statistically significantly associated with disease-specific survival in the full case analysis or in the stage-stratified analyses. Three SNPs were statistically significantly associated with disease-specific survival for cases with tumors located in the distal colon (rs698022, HR = 1.48, CI 1.30-1.69, p = 8.47 × 10) and the proximal colon (rs189655236, HR = 2.14, 95% CI 1.65-2.77, p = 9.19 × 10 and rs144717887, HR = 2.01, 95% CI 1.57-2.58, p = 3.14 × 10), whereas no associations were detected for rectal tumors. Findings from this large genome-wide association study highlight the potential for anatomical-site-stratified genome-wide studies to identify germline genetic risk variants associated with colorectal cancer-specific survival. Larger sample sizes and further replication efforts are needed to more fully interpret these findings.
新的遗传标记的鉴定可能会改善结直肠癌预后的预测。我们的目的是在对 16964 例结直肠癌病例的分析中,研究种系遗传变异与疾病特异性生存的全基因组关联。我们分析了来自 15 项研究的联盟的基因型和结直肠癌特异性生存数据。使用 Cox 比例风险模型,根据临床因素和主要成分,在对数加性模型下对大约 750 万个 SNP 进行了检验。此外,我们还进行了按肿瘤部位和疾病阶段分层的二次分析。我们使用全基因组 p 值阈值为 5×10 来评估统计学意义。在全病例分析或分期分层分析中,没有变异与疾病特异性生存具有统计学显著相关性。三个 SNP 与位于远端结肠(rs698022,HR=1.48,CI 1.30-1.69,p=8.47×10)和近端结肠(rs189655236,HR=2.14,95%CI 1.65-2.77,p=9.19×10 和 rs144717887,HR=2.01,95%CI 1.57-2.58,p=3.14×10)的病例的疾病特异性生存具有统计学显著相关性,而直肠肿瘤则没有相关性。这项大型全基因组关联研究的结果强调了基于解剖部位分层的全基因组研究识别与结直肠癌特异性生存相关的种系遗传风险变异的潜力。需要更大的样本量和进一步的复制工作来更全面地解释这些发现。