Programme in Cancer and Stem Cell Biology, Duke-NUS Medical School, Singapore, 169857, Singapore.
Centre for Computational Biology, Duke-NUS Medical School, Singapore, 169857, Singapore.
Sci Rep. 2022 Jan 10;12(1):390. doi: 10.1038/s41598-021-04207-6.
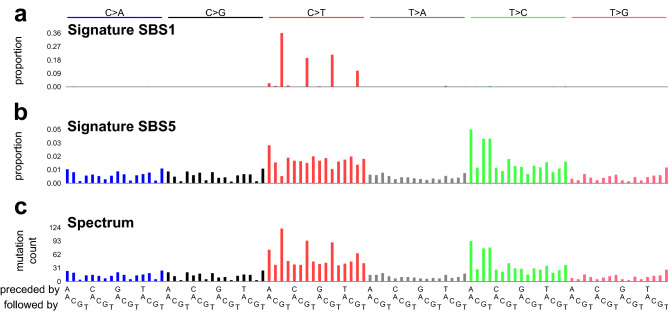
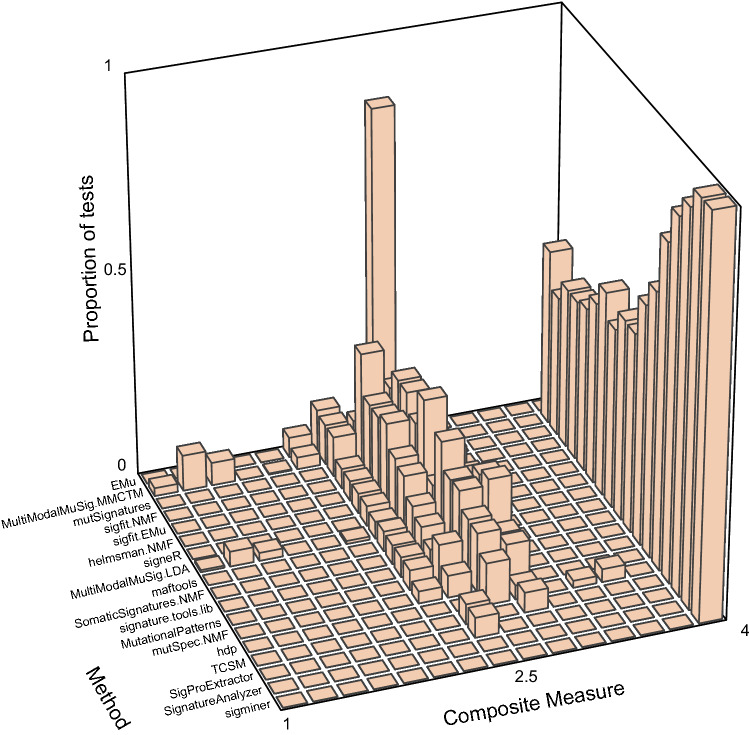
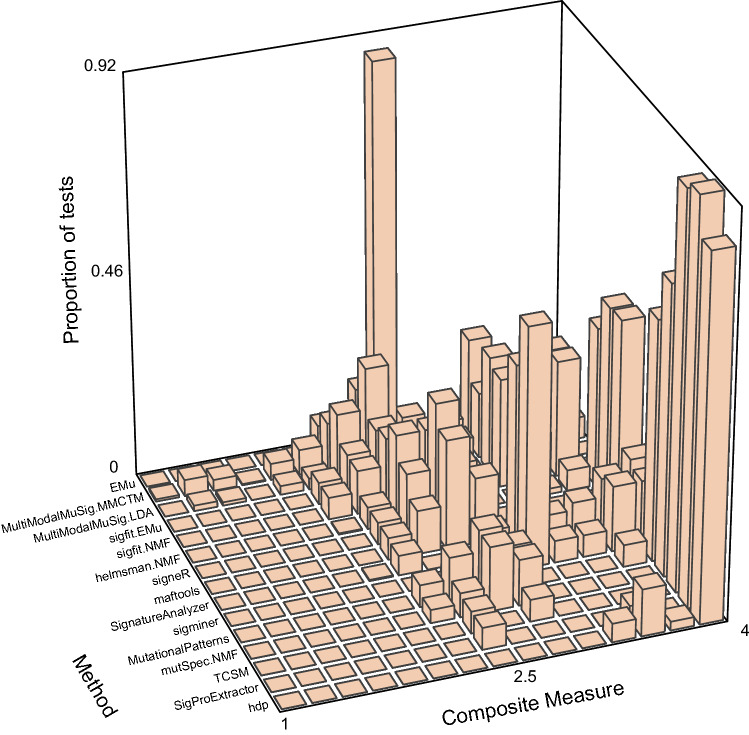
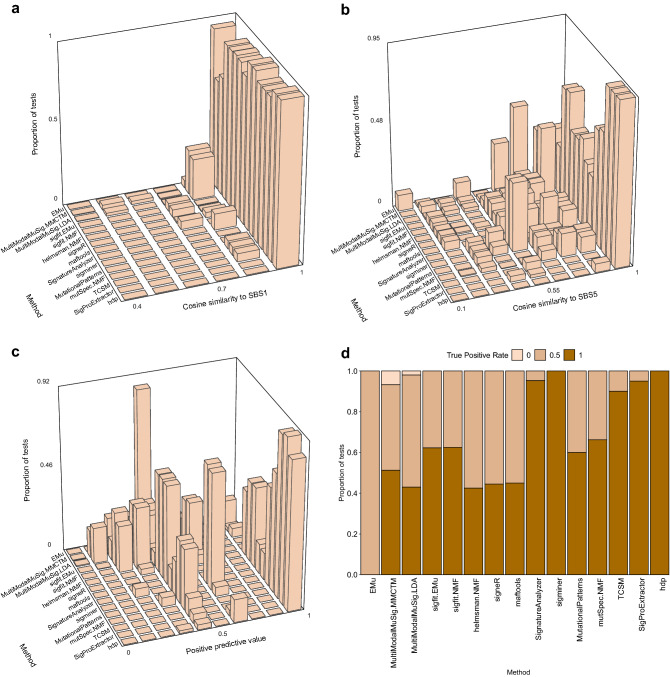
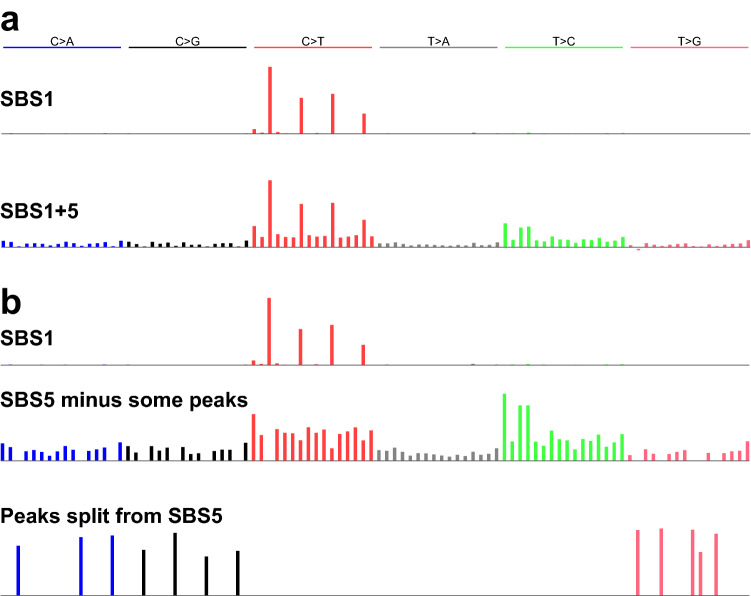
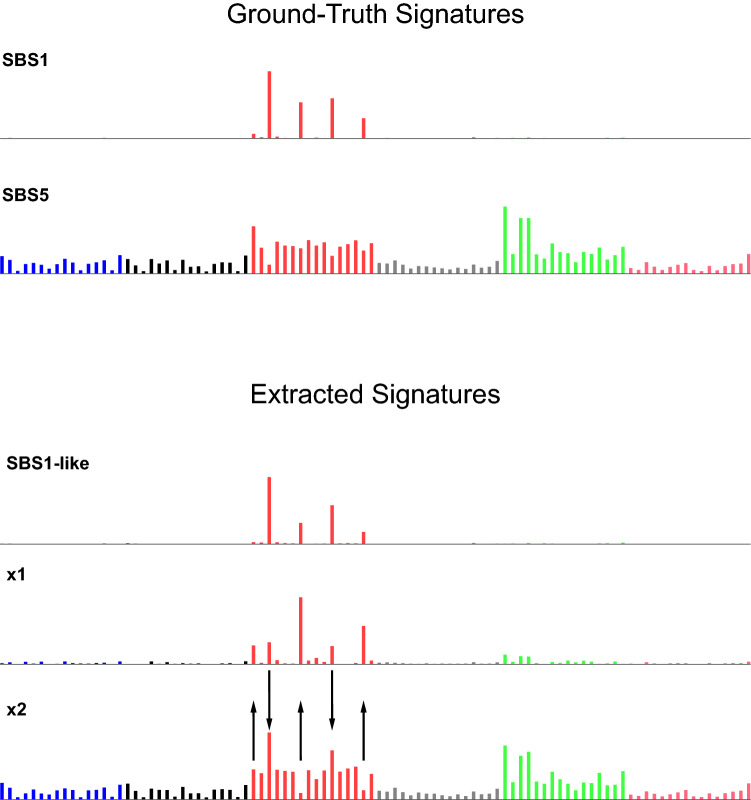
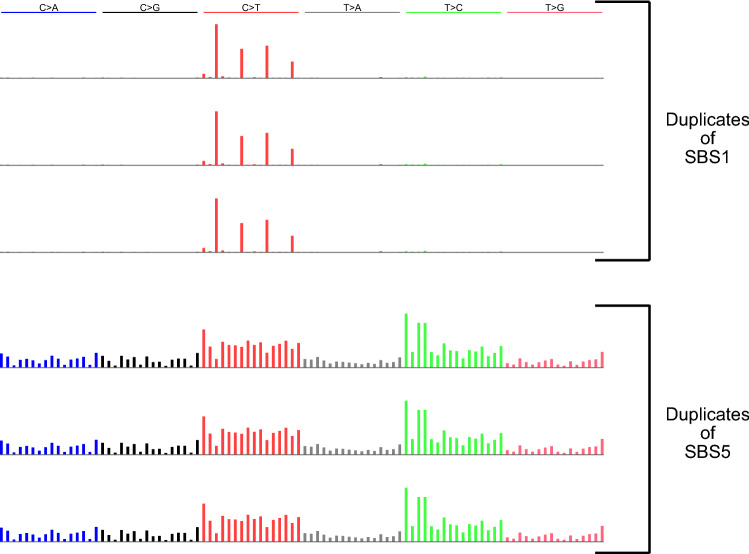
Mutational signatures are characteristic patterns of mutations generated by exogenous mutagens or by endogenous mutational processes. Mutational signatures are important for research into DNA damage and repair, aging, cancer biology, genetic toxicology, and epidemiology. Unsupervised learning can infer mutational signatures from the somatic mutations in large numbers of tumors, and separating correlated signatures is a notable challenge for this task. To investigate which methods can best meet this challenge, we assessed 18 computational methods for inferring mutational signatures on 20 synthetic data sets that incorporated varying degrees of correlated activity of two common mutational signatures. Performance varied widely, and four methods noticeably outperformed the others: hdp (based on hierarchical Dirichlet processes), SigProExtractor (based on multiple non-negative matrix factorizations over resampled data), TCSM (based on an approach used in document topic analysis), and mutSpec.NMF (also based on non-negative matrix factorization). The results underscored the complexities of mutational signature extraction, including the importance and difficulty of determining the correct number of signatures and the importance of hyperparameters. Our findings indicate directions for improvement of the software and show a need for care when interpreting results from any of these methods, including the need for assessing sensitivity of the results to input parameters.
突变特征是由外源性诱变剂或内源性突变过程产生的突变的特征模式。突变特征对于研究 DNA 损伤和修复、衰老、癌症生物学、遗传毒理学和流行病学非常重要。无监督学习可以从大量肿瘤的体细胞突变中推断出突变特征,而分离相关特征是该任务的一个显著挑战。为了研究哪些方法可以最好地应对这一挑战,我们评估了 18 种计算方法,这些方法用于推断 20 个合成数据集上的突变特征,这些数据集包含两种常见突变特征的相关活动的不同程度。性能差异很大,有四种方法明显优于其他方法:hdp(基于层次狄利克雷过程)、SigProExtractor(基于对重采样数据的多次非负矩阵分解)、TCSM(基于文档主题分析中使用的方法)和 mutSpec.NMF(也基于非负矩阵分解)。结果强调了突变特征提取的复杂性,包括确定正确特征数量的重要性和难度,以及超参数的重要性。我们的研究结果为软件的改进指明了方向,并表明在解释这些方法中的任何一种方法的结果时都需要谨慎,包括需要评估结果对输入参数的敏感性。