Institute of Clinical Genetics and Tumor Genetics, Bonn, Germany.
Leibniz Institute for Analytical Sciences (ISAS), Dortmund, Germany.
Orphanet J Rare Dis. 2022 Jan 31;17(1):29. doi: 10.1186/s13023-021-02068-w.
Goltz syndrome (GS) is a X-linked disorder defined by defects of mesodermal- and ectodermal-derived structures and caused by PORCN mutations. Features include striated skin-pigmentation, ocular and skeletal malformations and supernumerary or hypoplastic nipples. Generally, GS is associated with in utero lethality in males and most of the reported male patients show mosaicism (only three non-mosaic surviving males have been described so far). Also, precise descriptions of neurological deficits in GS are rare and less severe phenotypes might not only be caused by mosaicism but also by less pathogenic mutations suggesting the need of a molecular genetics and functional work-up of these rare variants.
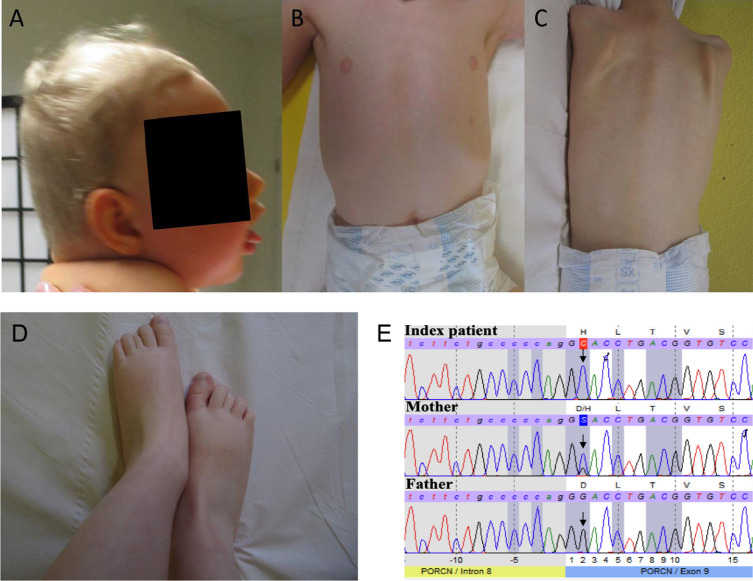
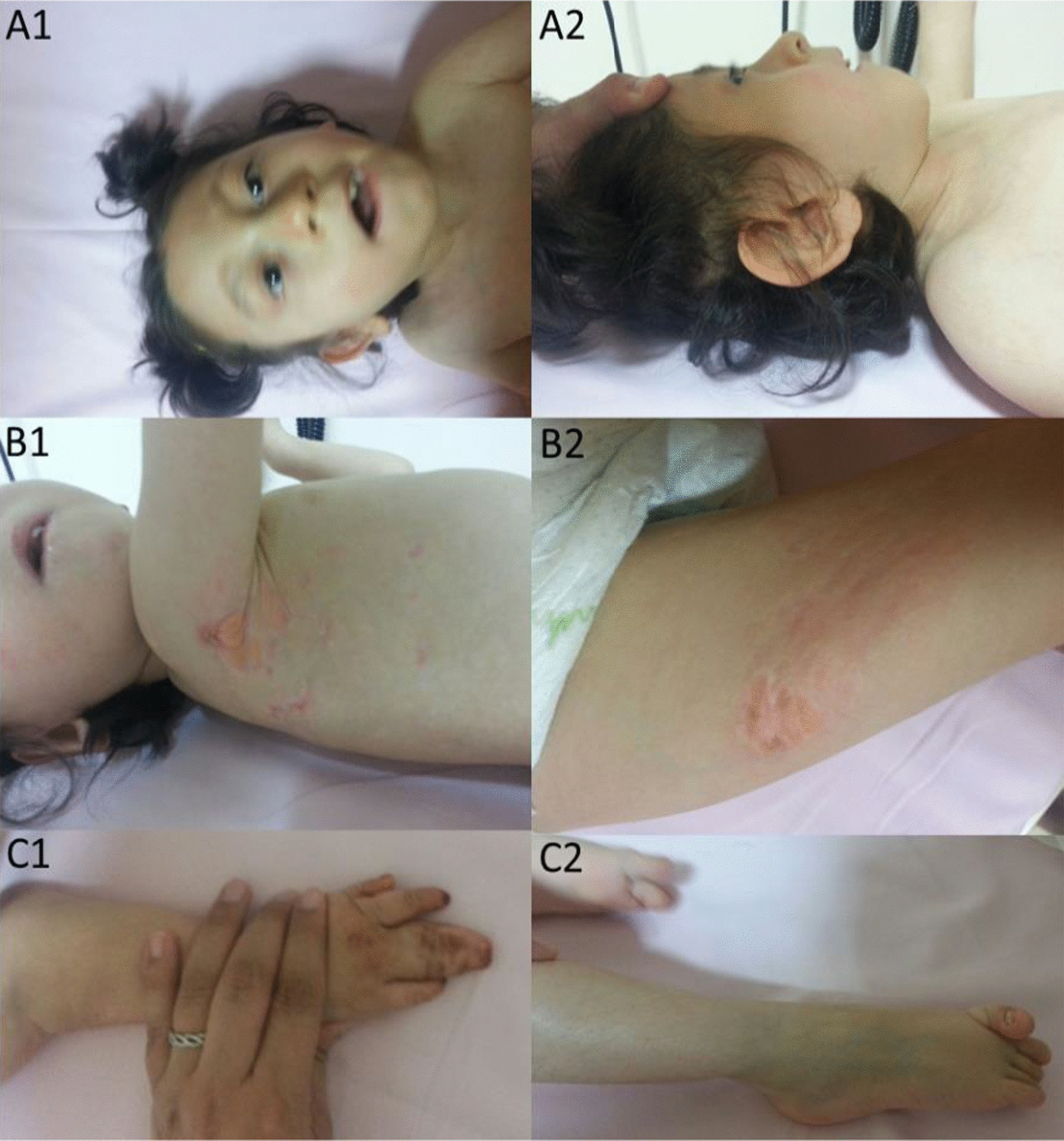
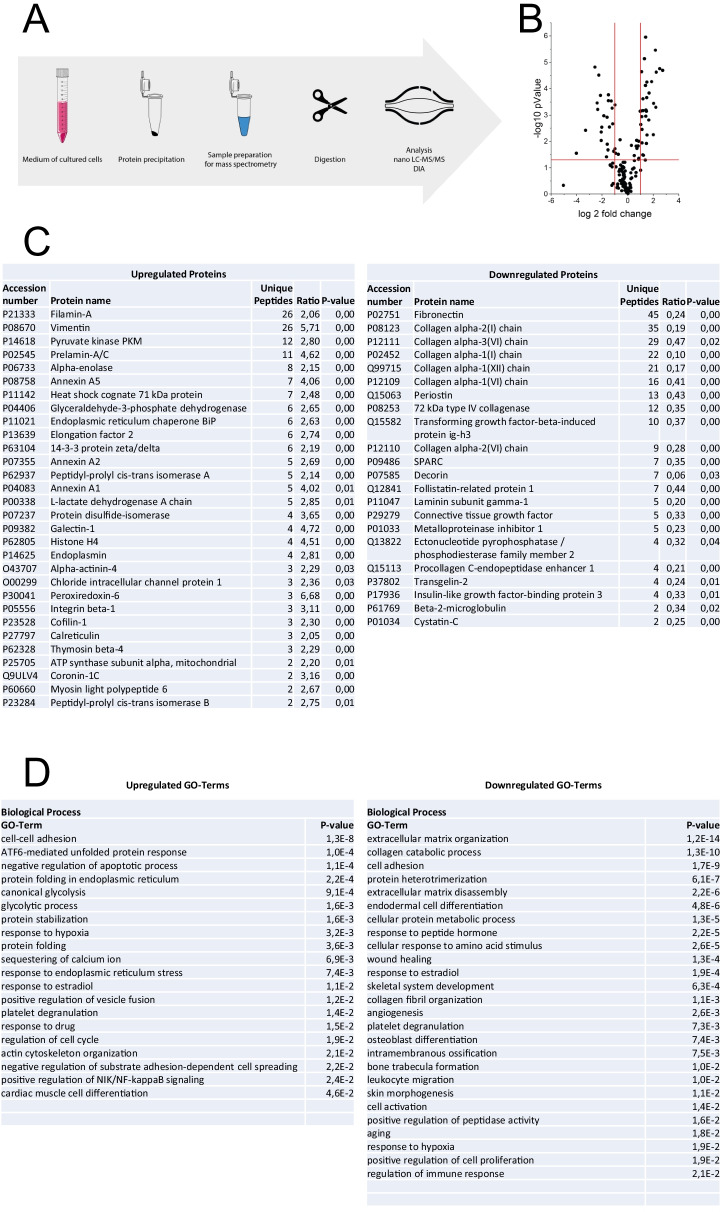
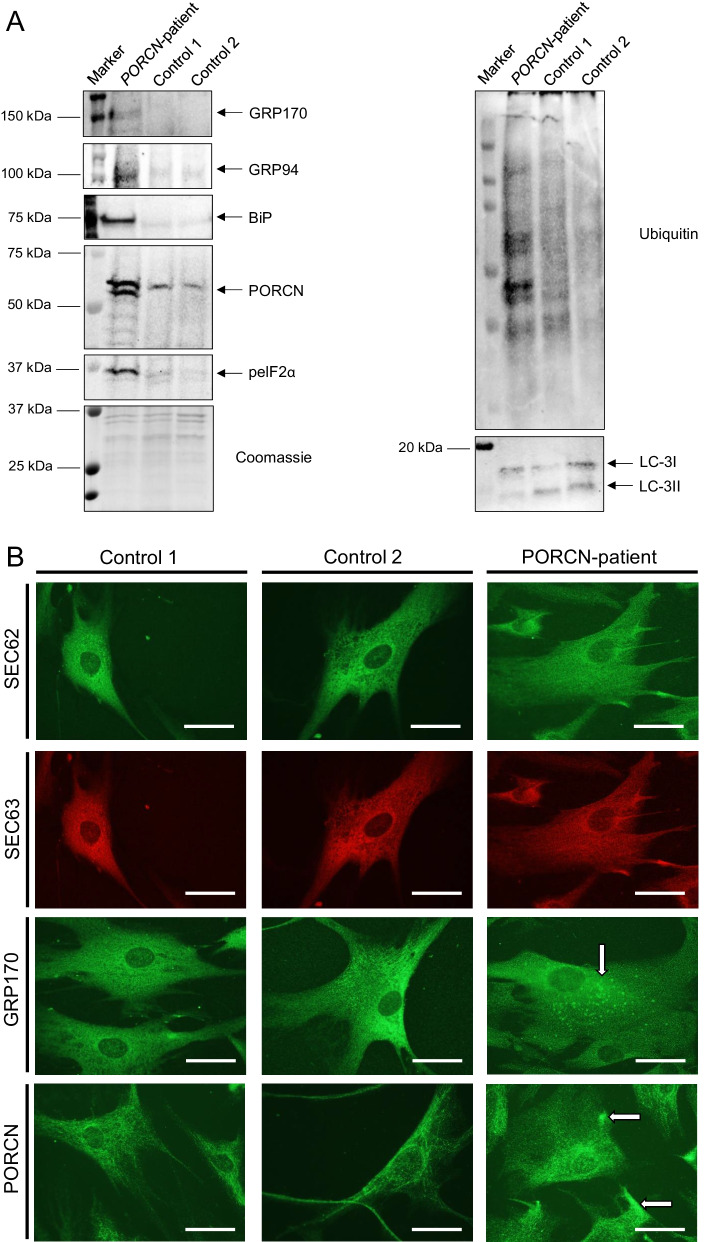
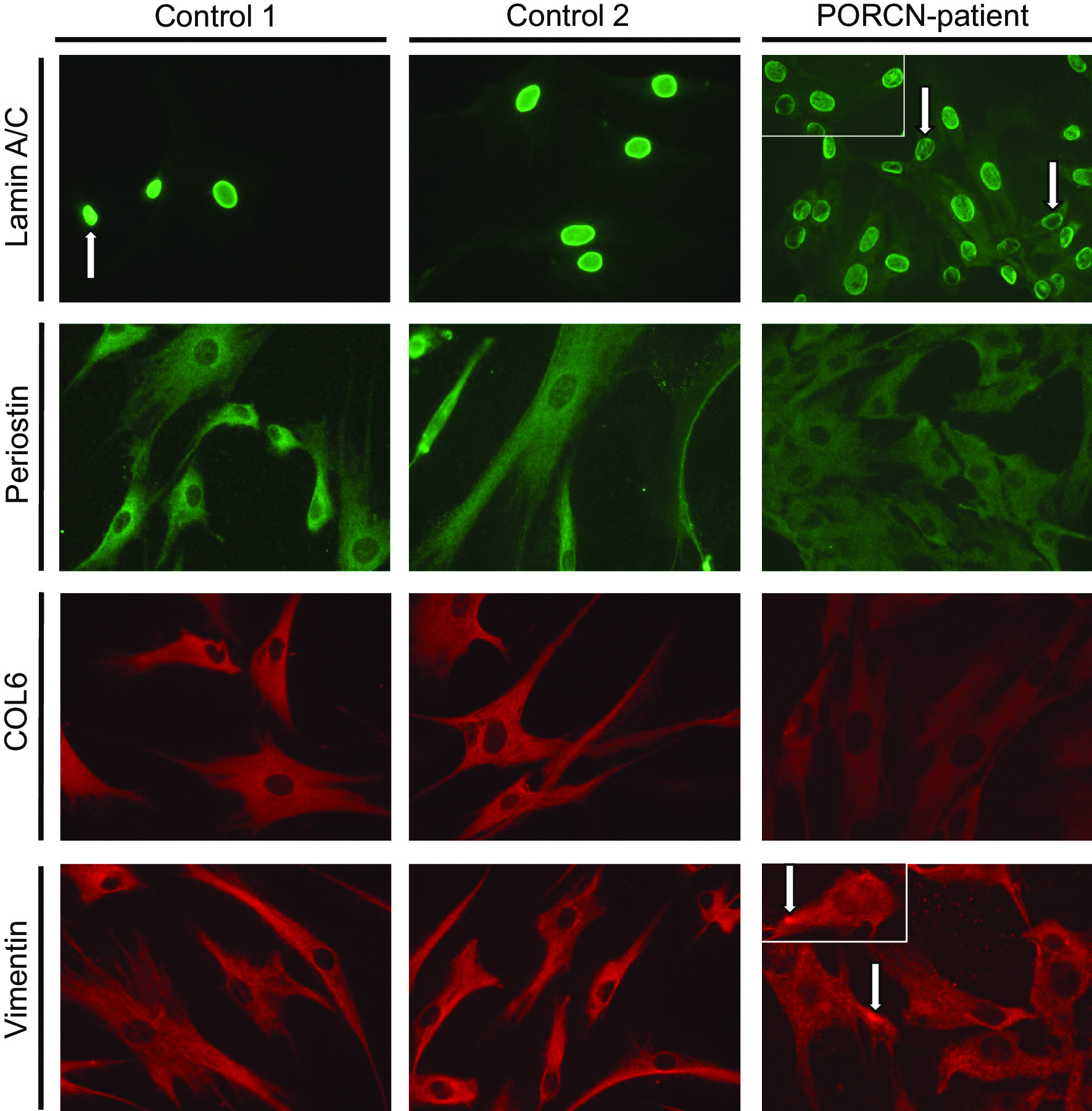
We report two cases: one girl suffering from typical skin and skeletal abnormalities, developmental delay, microcephaly, thin corpus callosum, periventricular gliosis and drug-resistant epilepsy caused by a PORCN nonsense-mutation (c.283C > T, p.Arg95Ter). Presence of these combined neurological features indicates that CNS-vulnerability might be a guiding symptom in the diagnosis of GS patients. The other patient is a boy with a supernumerary nipple and skeletal anomalies but also, developmental delay, microcephaly, cerebral atrophy with delayed myelination and drug-resistant epilepsy as predominant features. Skin abnormalities were not observed. Genotyping revealed a novel PORCN missense-mutation (c.847G > C, p.Asp283His) absent in the Genome Aggregation Database (gnomAD) but also identified in his asymptomatic mother. Given that non-random X-chromosome inactivation was excluded in the mother, fibroblasts of the index had been analyzed for PORCN protein-abundance and -distribution, vulnerability against additional ER-stress burden as well as for protein secretion revealing changes.
Our combined findings may suggest incomplete penetrance for the p.Asp283His variant and provide novel insights into the molecular etiology of GS by adding impaired ER-function and altered protein secretion to the list of pathophysiological processes resulting in the clinical manifestation of GS.
戈尔茨综合征(GS)是一种 X 连锁疾病,由中胚层和外胚层来源的结构缺陷引起,由 PORCN 突变引起。其特征包括条纹状皮肤色素沉着、眼和骨骼畸形以及多余或发育不全的乳头。通常,GS 会导致男性宫内死亡,大多数报道的男性患者表现为嵌合体(迄今为止仅描述了三名非嵌合存活的男性)。此外,GS 中神经缺陷的精确描述很少见,较轻的表型不仅可能是由嵌合体引起的,也可能是由致病性较低的突变引起的,这表明需要对这些罕见变体进行分子遗传学和功能分析。
我们报告了两个病例:一个女孩患有典型的皮肤和骨骼异常、发育迟缓、小头畸形、薄胼胝体、脑室周围神经胶质增生和耐药性癫痫,这是由 PORCN 无意义突变(c.283C>T,p.Arg95Ter)引起的。这些联合神经特征的存在表明,中枢神经系统易损性可能是 GS 患者诊断的一个指导症状。另一个患者是一个男孩,他有多余的乳头和骨骼异常,但也有发育迟缓、小头畸形、脑萎缩伴髓鞘化延迟和耐药性癫痫作为主要特征。未观察到皮肤异常。基因分型显示 PORCN 错义突变(c.847G>C,p.Asp283His),该突变不存在于基因组聚集数据库(gnomAD)中,但在无症状的母亲中也发现了该突变。由于排除了母亲的非随机 X 染色体失活,我们分析了该指数的成纤维细胞的 PORCN 蛋白丰度和分布、对额外 ER 应激负担的脆弱性以及蛋白分泌,发现了变化。
我们的综合发现可能表明 p.Asp283His 变体的不完全外显率,并通过将 ER 功能障碍和改变的蛋白分泌添加到导致 GS 临床表现的病理生理过程列表中,为 GS 的分子病因提供新的见解。