Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Key Laboratory of Molecular Cardiovascular Science, Ministry of Education, Peking University, 38 Xueyuan Road, Haidian District, Beijing 100191, China.
Department of Interventional Radiology and Vascular Surgery, Peking University Third Hospital, North Garden Road 49, Haidian District, Beijing 100191, China.
Redox Biol. 2022 Apr;50:102257. doi: 10.1016/j.redox.2022.102257. Epub 2022 Feb 4.
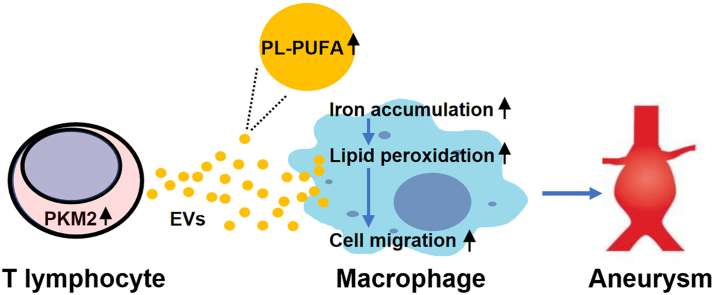
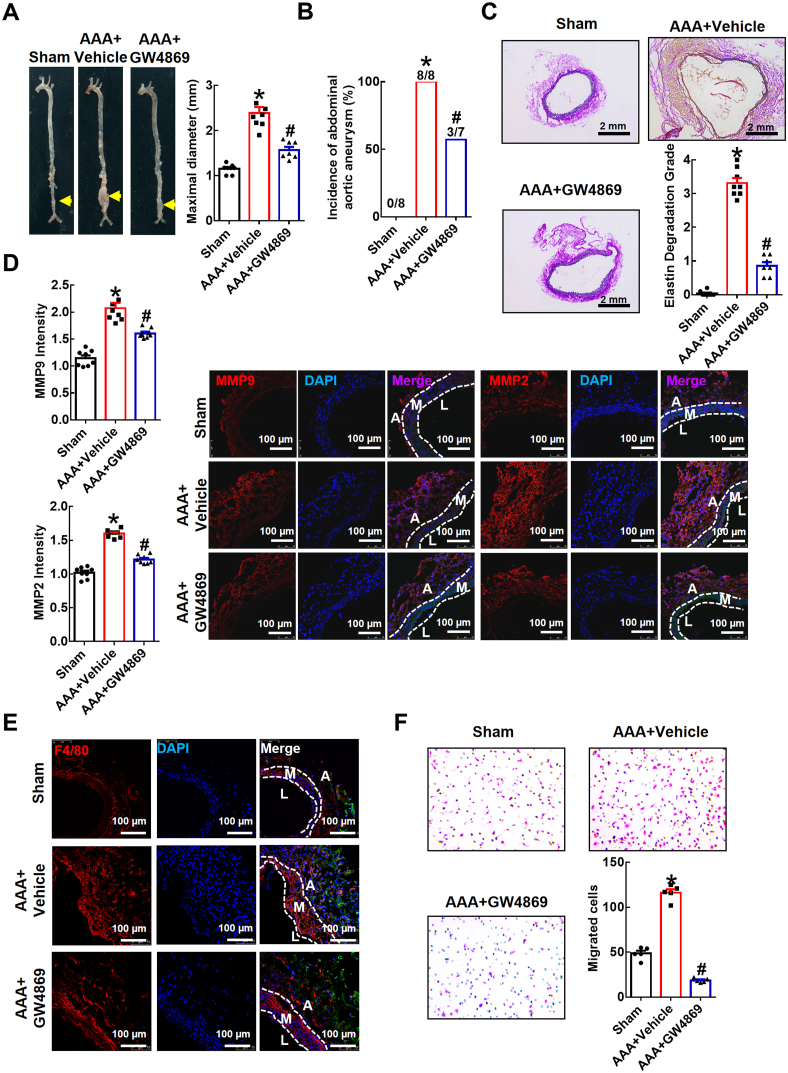
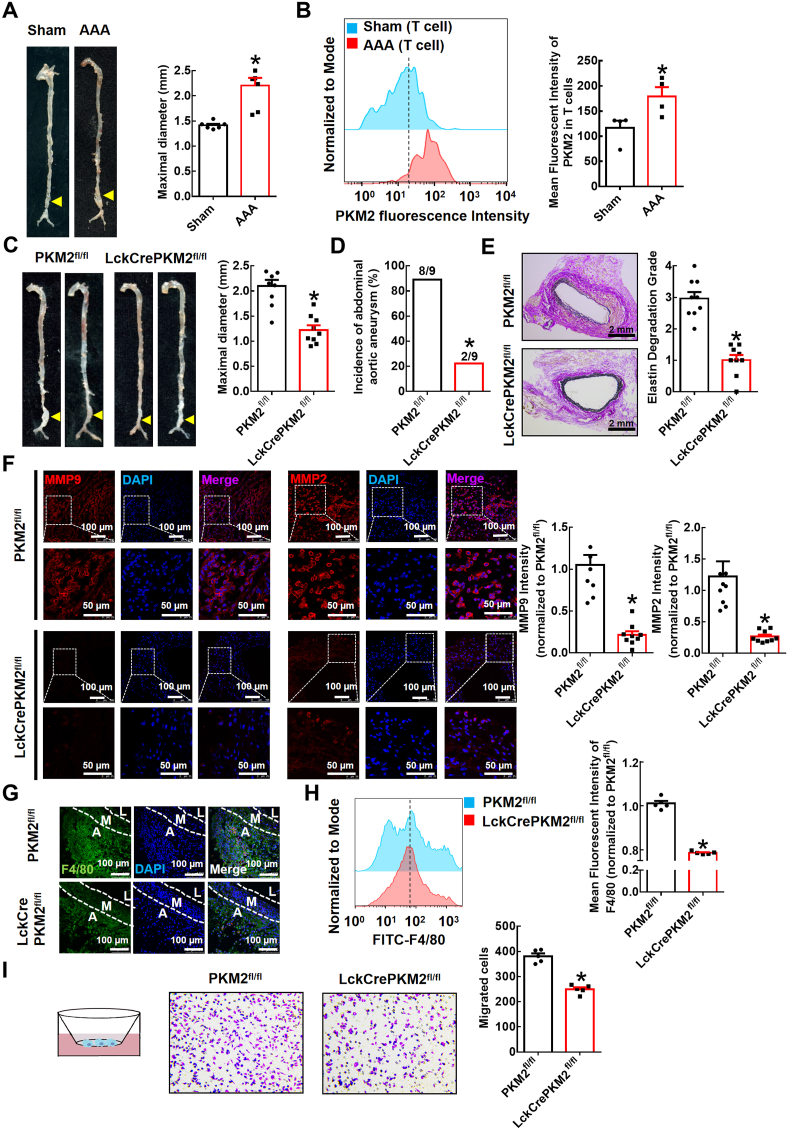
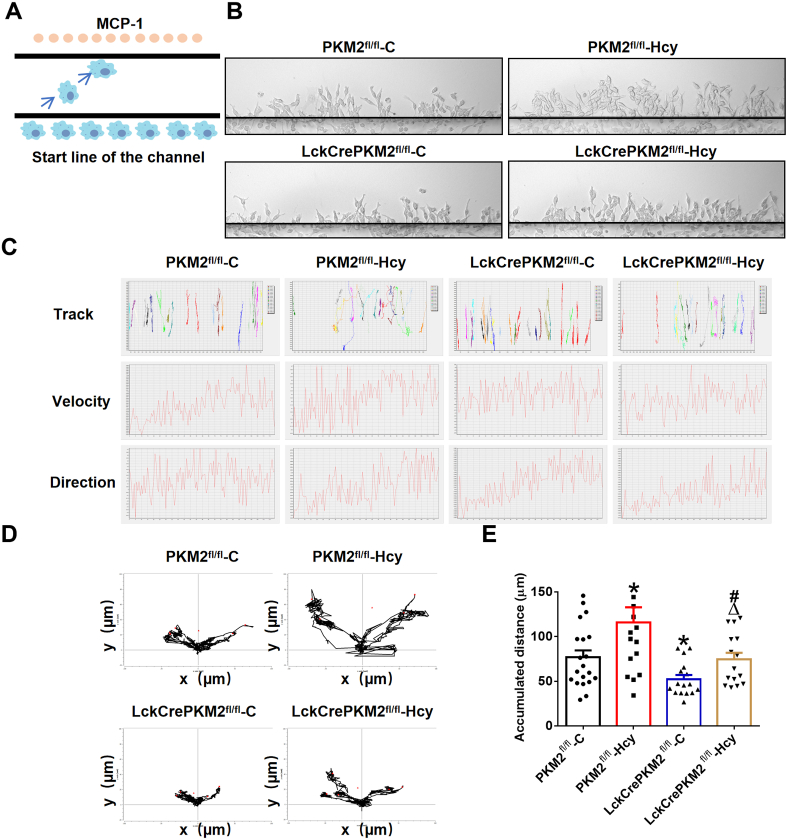
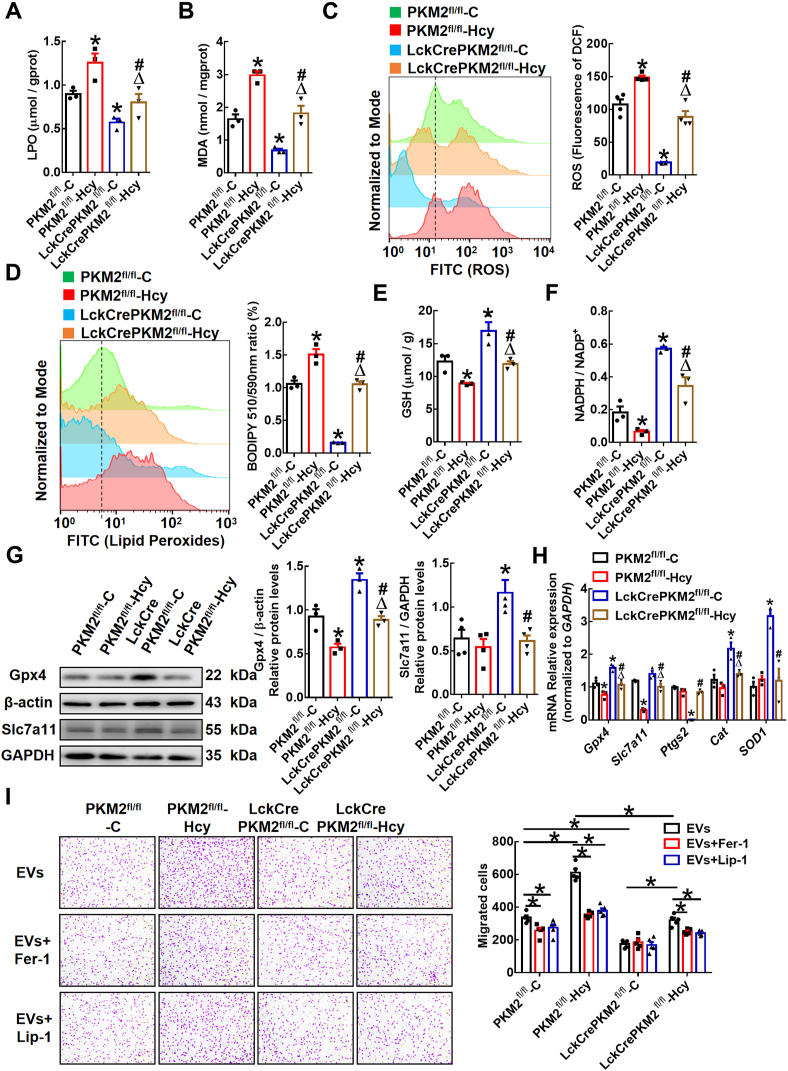
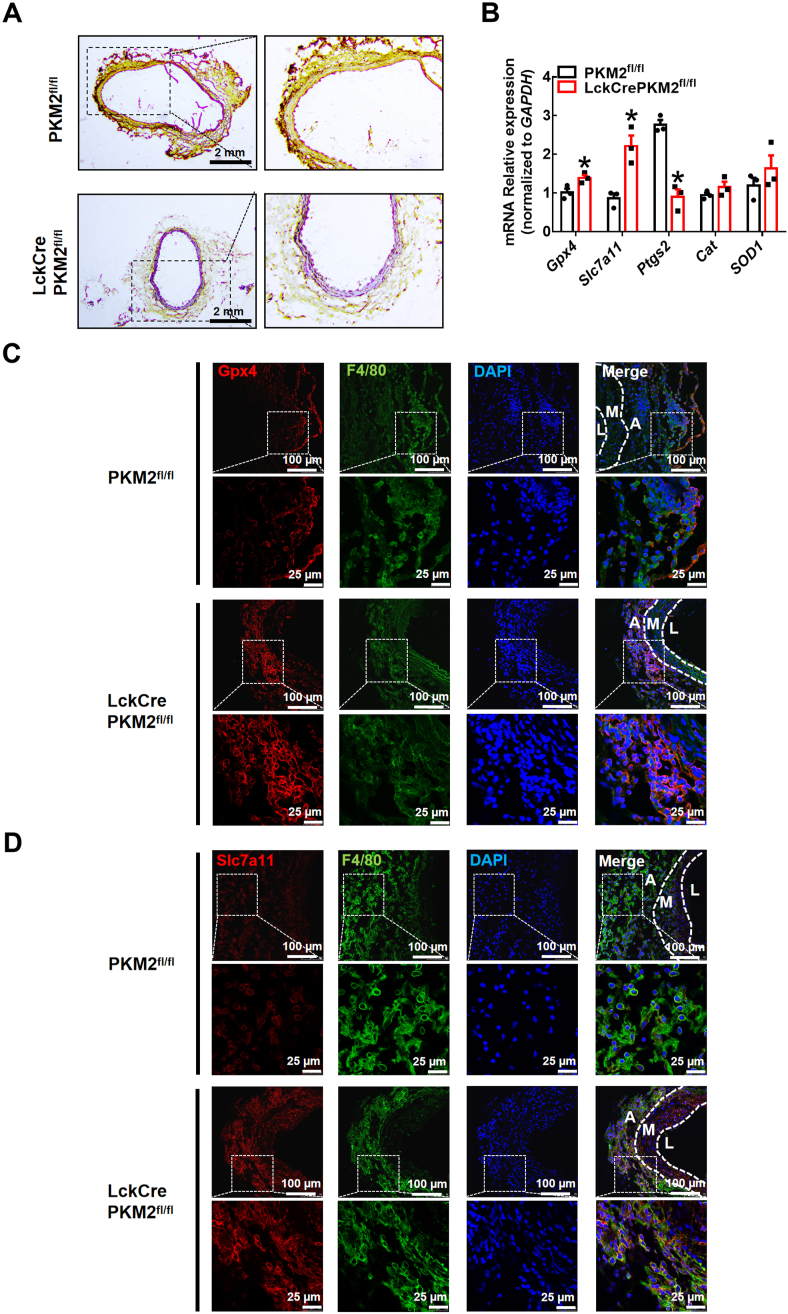
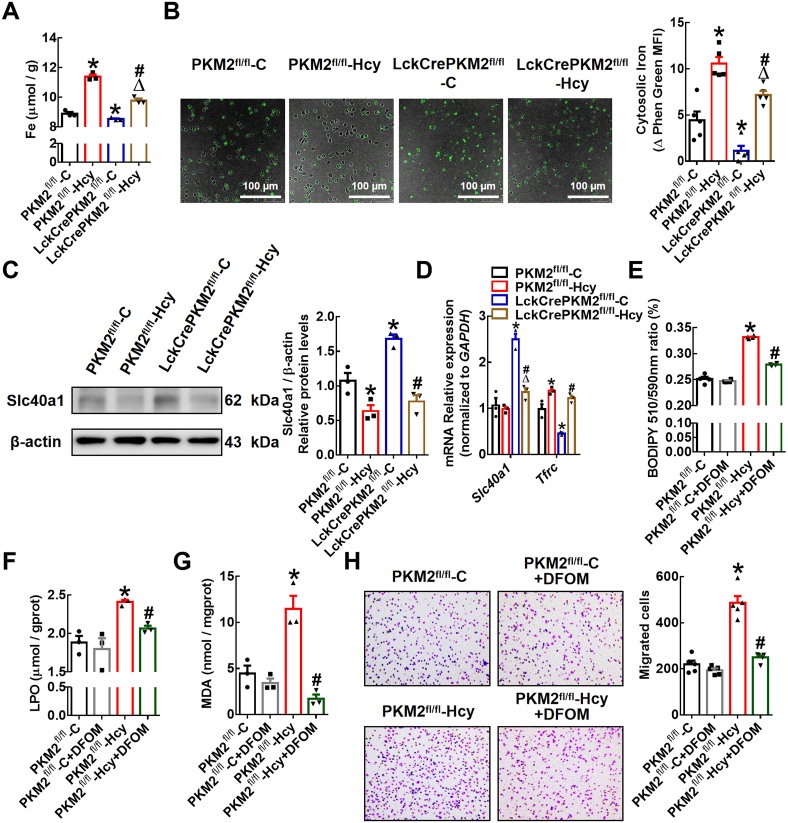
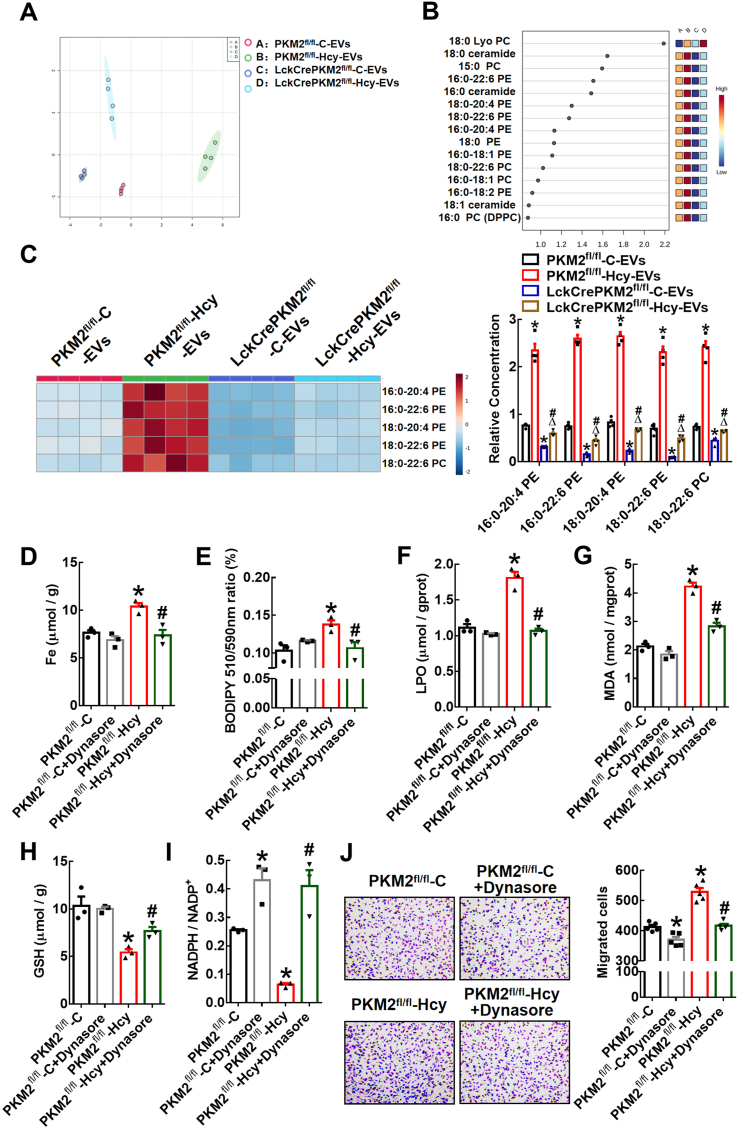
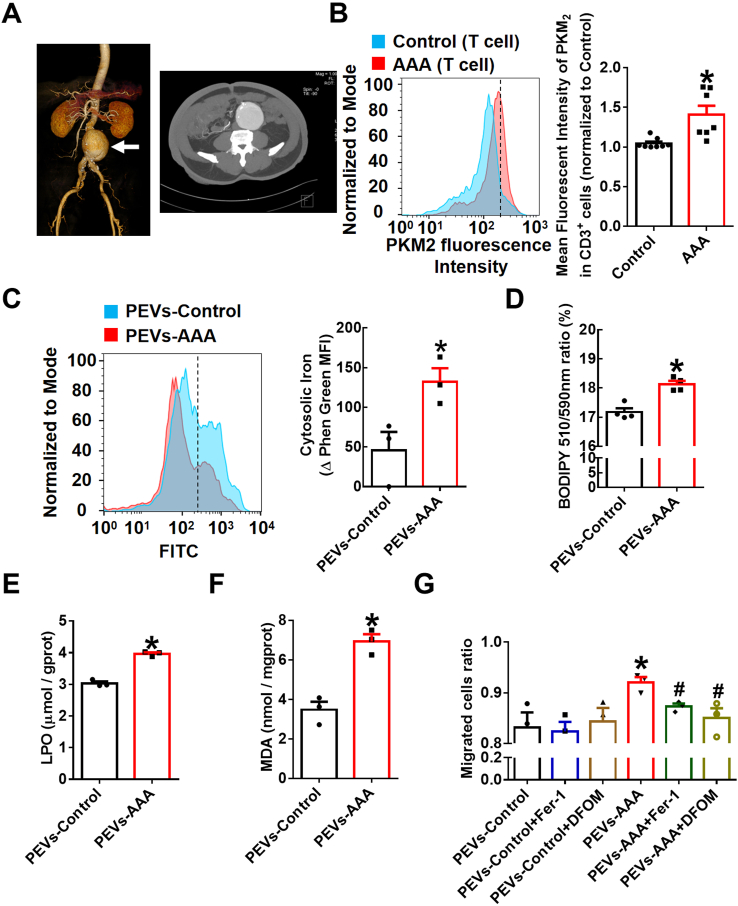
T lymphocyte and macrophage infiltration in the aortic wall is critical for abdominal aortic aneurysm (AAA). However, how T lymphocytes interact with macrophages in the pathogenesis of AAA remains largely uncharacterized. In an elastase-induced murine AAA model, we first found that the expression of pyruvate kinase muscle isozyme 2 (PKM2), the last rate-limiting enzyme in glycolysis, was increased in infiltrated T lymphocytes of vascular lesions. T lymphocyte-specific PKM2 deficiency in mice (LckCrePKM2) or intraperitoneal administration of the sphingomyelinase inhibitor GW4869 caused a significant attenuation of the elastase-increased aortic diameter, AAA incidence, elastic fiber disruption, matrix metalloproteinases (MMPs) expression, and macrophage infiltration in the vascular adventitia compared with those in PKM2 mice. Mechanistically, extracellular vesicles (EVs) derived from PKM2-activated T lymphocytes elevated macrophage iron accumulation, lipid peroxidation, and migration in vitro, while macrophages treated with EVs from PKM2-null T lymphocytes or pretreated with the lipid peroxidation inhibitors ferrostatin-1 (Fer-1), liproxstatin-1 (Lip-1), or the iron chelating agent deferoxamine mesylate (DFOM) reversed these effects. In vascular lesions of elastase-induced LckCrePKM2 mice with AAA, the oxidant system weakened, with downregulated 4-hydroxynonenal (4-HNE) levels and strengthened antioxidant defense systems with upregulated glutathione peroxidase 4 (GPX4) and cystine/glutamate antiporter solute carrier family 7 member 11 (Slc7a11) expressions in macrophages. High-throughput metabolomics showed that EVs derived from PKM2-activated T lymphocytes contained increased levels of polyunsaturated fatty acid (PUFA)-containing phospholipids, which may provide abundant substrates for lipid peroxidation in target macrophages. More importantly, upregulated T lymphocyte PKM2 expression was also found in clinical AAA subjects, and EVs isolated from AAA patient plasma enhanced macrophage iron accumulation, lipid peroxidation, and migration ex vivo. Therefore, from cell-cell crosstalk and metabolic perspectives, the present study shows that PKM2-activated T lymphocyte-derived EVs may drive AAA progression by promoting macrophage redox imbalance and migration, and targeting the T lymphocyte-EV-macrophage axis may be a potential strategy for early warning and treating AAA.
T 淋巴细胞和巨噬细胞浸润在主动脉壁中对于腹主动脉瘤(AAA)至关重要。然而,T 淋巴细胞如何与巨噬细胞相互作用在 AAA 的发病机制中仍很大程度上尚未被描述。在弹性蛋白酶诱导的小鼠 AAA 模型中,我们首先发现,糖酵解的最后限速酶丙酮酸激酶肌肉同工酶 2(PKM2)的表达在血管病变浸润的 T 淋巴细胞中增加。在小鼠中特异性敲除 T 淋巴细胞中的 PKM2(LckCrePKM2)或腹腔内给予鞘磷脂酶抑制剂 GW4869,与 PKM2 小鼠相比,弹性蛋白酶引起的主动脉直径增加、AAA 发生率、弹性纤维破坏、基质金属蛋白酶(MMPs)表达和血管外膜巨噬细胞浸润明显减弱。在机制上,来自 PKM2 激活的 T 淋巴细胞的细胞外囊泡(EVs)在体外升高了巨噬细胞的铁积累、脂质过氧化和迁移,而用来自 PKM2 缺失的 T 淋巴细胞的 EVs 处理的巨噬细胞或用脂质过氧化抑制剂 ferrostatin-1(Fer-1)、liproxstatin-1(Lip-1)或铁螯合剂甲磺酸去铁胺(DFOM)预处理的巨噬细胞则逆转了这些作用。在弹性蛋白酶诱导的具有 AAA 的 LckCrePKM2 小鼠的血管病变中,氧化应激系统减弱,4-羟基壬烯醛(4-HNE)水平下调,谷胱甘肽过氧化物酶 4(GPX4)和胱氨酸/谷氨酸反向转运蛋白溶质载体家族 7 成员 11(Slc7a11)的表达增强,抗氧化防御系统增强。高通量代谢组学显示,来自 PKM2 激活的 T 淋巴细胞的 EVs 含有增加水平的多不饱和脂肪酸(PUFA)含磷酯,这可能为靶巨噬细胞中的脂质过氧化提供丰富的底物。更重要的是,在临床 AAA 患者中也发现上调的 T 淋巴细胞 PKM2 表达,从 AAA 患者血浆中分离的 EVs 增强了巨噬细胞的铁积累、脂质过氧化和迁移。因此,从细胞-细胞通讯和代谢的角度来看,本研究表明 PKM2 激活的 T 淋巴细胞衍生的 EVs 可能通过促进巨噬细胞氧化还原失衡和迁移来驱动 AAA 的进展,靶向 T 淋巴细胞-EV-巨噬细胞轴可能是早期预警和治疗 AAA 的潜在策略。