Hata Hiroaki, Phuoc Tran Duy, Marzouk Sobeh Mohamed, Kitao Akio
School of Life Science and Technology, Tokyo Institute of Technology, Meguro-ku, Tokyo 152-8550, Japan.
Physics Department, Faculty of Science, Ain Shams University, Cairo 11566, Egypt.
Biophys Physicobiol. 2021 Dec 4;18:305-316. doi: 10.2142/biophysico.bppb-v18.037. eCollection 2021.
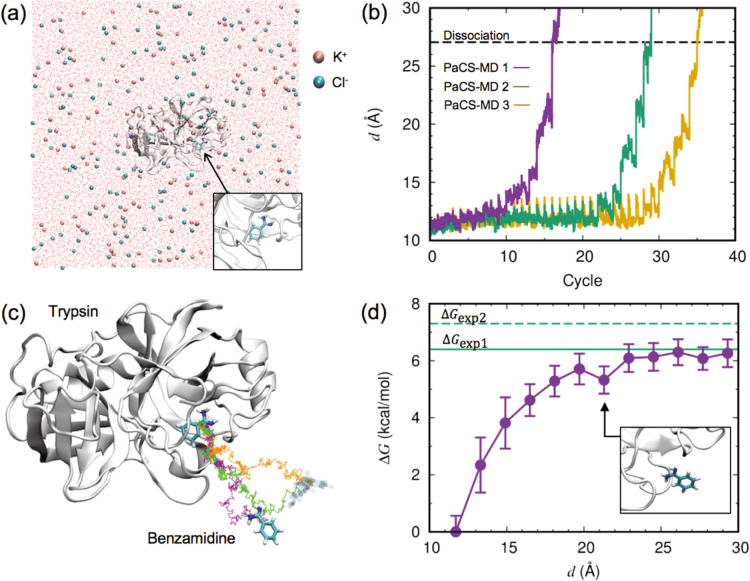
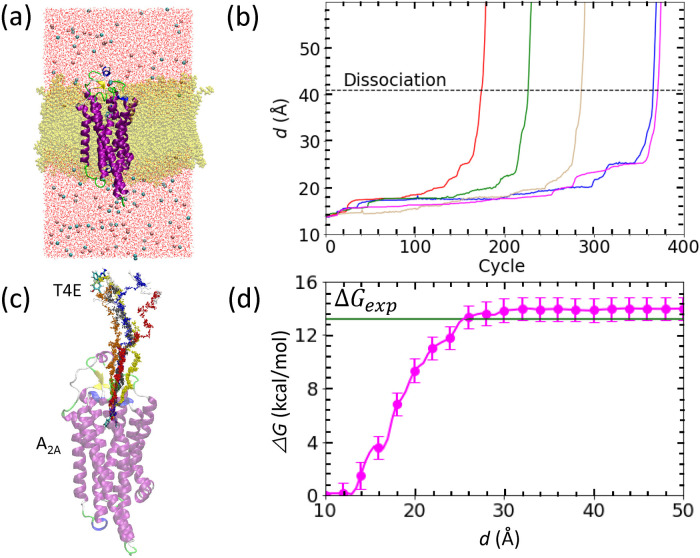
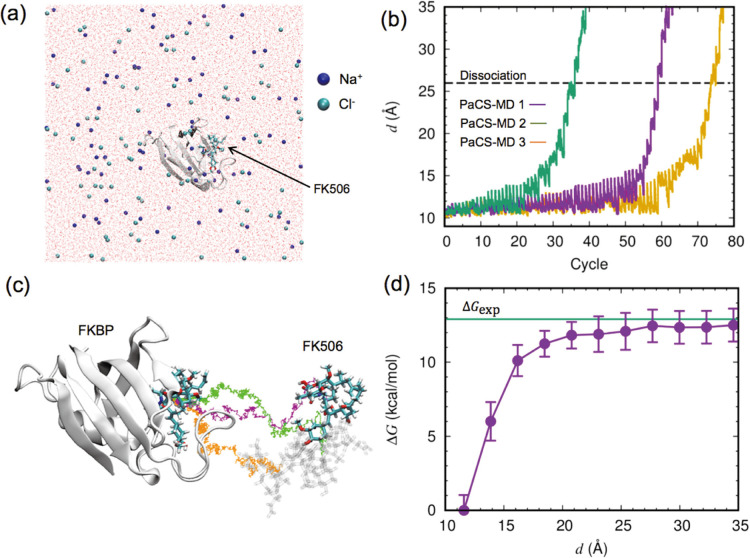
We recently proposed a computational procedure to simulate the dissociation of protein/ligand complexes using the dissociation Parallel Cascade Selection Molecular Dynamics simulation (dPaCS-MD) method and to analyze the generated trajectories using the Markov state model (MSM). This procedure, called dPaCS-MD/MSM, enables calculation of the dissociation free energy profile and the standard binding free energy. To examine whether this method can reproduce experimentally determined binding free energies for a variety of systems, we used it to investigate the dissociation of three protein/ligand complexes: trypsin/benzamine, FKBP/FK506, and adenosine A receptor/T4E. First, dPaCS-MD generated multiple dissociation pathways within a reasonable computational time for all the complexes, although the complexes differed significantly in the size of the molecules and in intermolecular interactions. Subsequent MSM analyses produced free energy profiles for the dissociations, which provided insights into how each ligand dissociates from the protein. The standard binding free energies obtained by dPaCS-MD/MSM are in good agreement with experimental values for all the complexes. We conclude that dPaCS-MD/MSM can accurately calculate the binding free energies of these complexes.
我们最近提出了一种计算程序,使用解离平行级联选择分子动力学模拟(dPaCS-MD)方法来模拟蛋白质/配体复合物的解离,并使用马尔可夫状态模型(MSM)分析生成的轨迹。这个程序称为dPaCS-MD/MSM,能够计算解离自由能分布和标准结合自由能。为了检验该方法是否能够重现各种系统实验测定的结合自由能,我们用它来研究三种蛋白质/配体复合物的解离:胰蛋白酶/苯胺、FKBP/FK506和腺苷A受体/T4E。首先,dPaCS-MD在合理的计算时间内为所有复合物生成了多条解离途径,尽管这些复合物在分子大小和分子间相互作用方面存在显著差异。随后的MSM分析产生了解离的自由能分布,这为每个配体如何从蛋白质上解离提供了见解。通过dPaCS-MD/MSM获得的标准结合自由能与所有复合物的实验值高度吻合。我们得出结论,dPaCS-MD/MSM能够准确计算这些复合物的结合自由能。