Department of Experimental and Clinical Medicine, "Magna Graecia" University of Catanzaro, Italy; Interdepartmental Center of Services (CIS), Molecular Genomics and Pathology, "Magna Græcia" University of Catanzaro, Italy.
Department of Health Sciences, "Magna Graecia" University of Catanzaro, Italy.
Infect Genet Evol. 2022 Apr;99:105253. doi: 10.1016/j.meegid.2022.105253. Epub 2022 Feb 18.
Nursing homes have represented important hotspots of viral spread during the initial wave of COVID-19 pandemics. The proximity of patients inside nursing homes allows investigate the dynamics of viral transmission, which may help understand SARS-Cov2 biology and spread.
SARS-CoV-2 viral genomes obtained from 46 patients infected in an outbreak inside a nursing home in Calabria region (South Italy) were analyzed by Next Generation Sequencing. We also investigated the evolution of viral genomes in 8 patients for which multiple swabs were available. Phylogenetic analysis and haplotype reconstruction were carried out with IQ-TREE software and RegressHaplo tool, respectively.
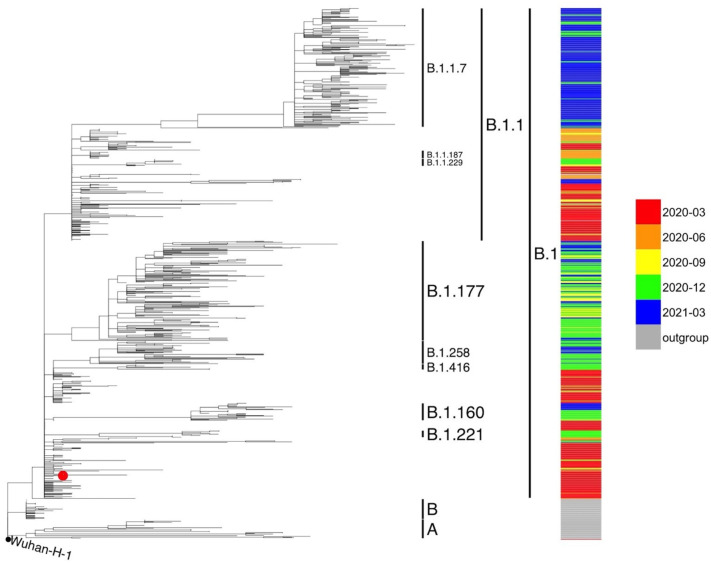
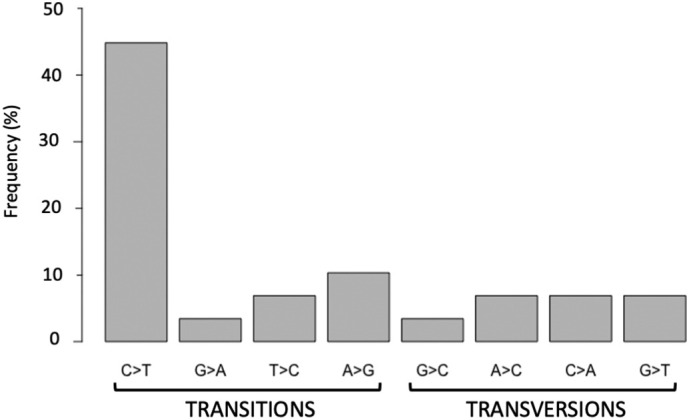
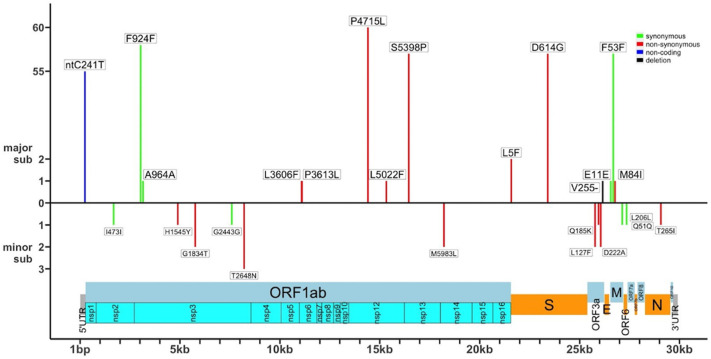
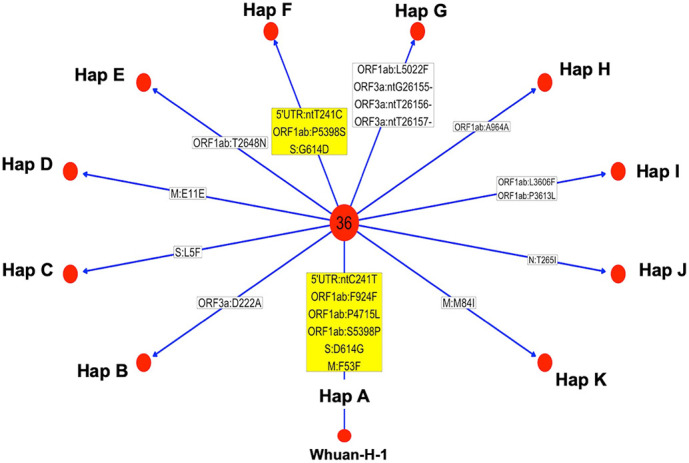
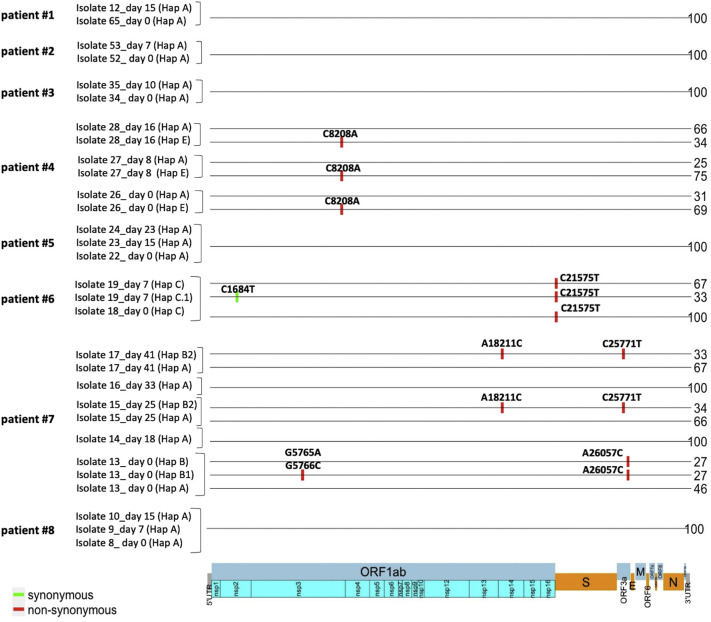
All viral strains isolated from patients infected in the nursing home were classified as B.1 lineage, clade G. Overall, 14 major single nucleotide variations (SNVs) (frequency > 80%) and 12 minor SNVs (frequency comprised between 20% and 80%) were identified with reference to the Wuhan-H-1 sequence (NC_045512.2). All patients presented the same 6 major SNVs: D614G in the S gene; P4715L, ntC3037T (F924F) and S5398P in Orf1ab gene; ntC26681T (F53F) in the M gene; and ntC241T in the non-coding UTR region. However, haplotype reconstruction identified a founder haplotype (Hap A) in 36 patients carrying only the 6 common SNVs indicated above, and 10 other haplotypes (Hap BK) derived from Hap A in the remaining 10 patients. Notably, no significant association between a specific viral haplotype and clinical parameters was found.
The predominant viral strain responsible for the infection in a nursing home in Calabria was the B.1 lineage (clade G). Viral genomes were classified into 11 haplotypes (Hap A in 36 patients, Hap BK in the remaining patients).
在 COVID-19 大流行的初始阶段,养老院是病毒传播的重要热点。养老院中患者之间的近距离接触使得研究病毒传播的动力学成为可能,这有助于了解 SARS-CoV-2 的生物学特性和传播方式。
通过下一代测序对来自意大利南部卡拉布里亚地区一家养老院爆发疫情中感染的 46 名患者的 SARS-CoV-2 病毒基因组进行分析。我们还对 8 名有多个拭子样本的患者的病毒基因组进行了进化研究。采用 IQ-TREE 软件进行系统发育分析和单倍型重建。
从养老院感染患者中分离的所有病毒株均归类为 B.1 谱系,G 分支。与武汉-H-1 序列(NC_045512.2)相比,总共鉴定出 14 个主要单核苷酸变异(SNV)(频率>80%)和 12 个次要 SNV(频率在 20%至 80%之间)。所有患者均存在相同的 6 个主要 SNV:S 基因中的 D614G;Orf1ab 基因中的 P4715L、ntC3037T(F924F)和 S5398P;M 基因中的 ntC26681T(F53F);非编码 UTR 区中的 ntC241T。然而,单倍型重建在 36 名仅携带上述 6 个常见 SNV 的患者中鉴定出一个起始单倍型(Hap A),在其余 10 名患者中从 Hap A 衍生出 10 个其他单倍型(Hap BK)。值得注意的是,未发现特定病毒单倍型与临床参数之间存在显著关联。
卡拉布里亚一家养老院感染的主要病毒株是 B.1 谱系(G 分支)。病毒基因组分为 11 个单倍型(36 名患者为 Hap A,其余患者为 Hap BK)。