School of Life Sciences, Arizona State University, Tempe, Arizona, United States of America.
Natural History Museum, Berlin, Germany.
PLoS Genet. 2022 Feb 24;18(2):e1010022. doi: 10.1371/journal.pgen.1010022. eCollection 2022 Feb.
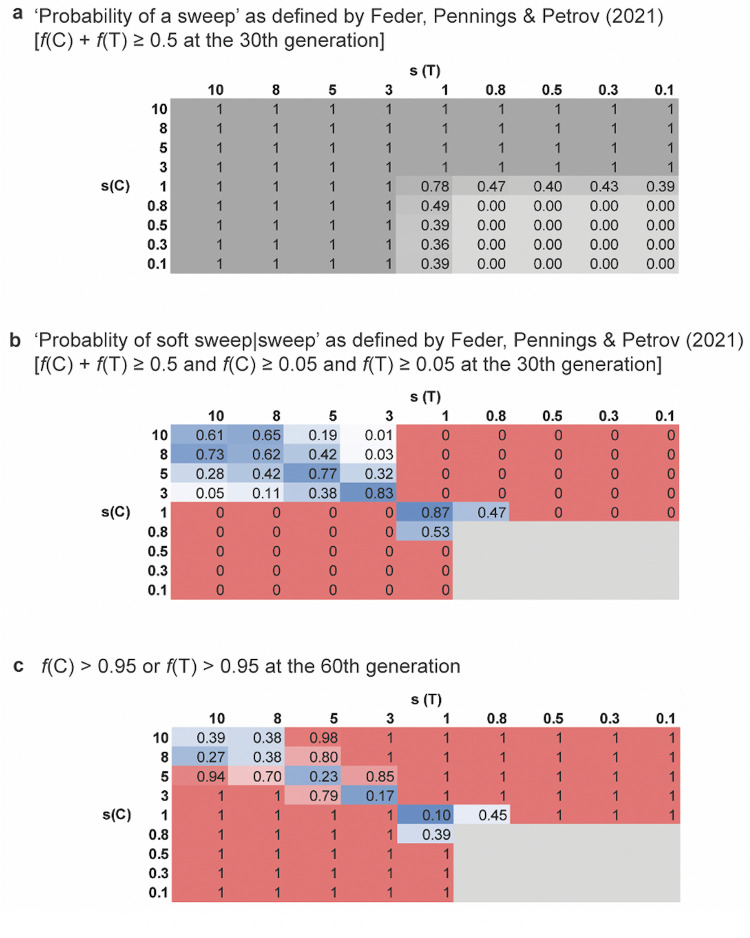
The ability to accurately identify and quantify genetic signatures associated with soft selective sweeps based on patterns of nucleotide variation has remained controversial. We here provide counter viewpoints to recent publications in PLOS Genetics that have argued not only for the statistical identifiability of soft selective sweeps, but also for their pervasive evolutionary role in both Drosophila and HIV populations. We present evidence that these claims owe to a lack of consideration of competing evolutionary models, unjustified interpretations of empirical outliers, as well as to new definitions of the processes themselves. Our results highlight the dangers of fitting evolutionary models based on hypothesized and episodic processes without properly first considering common processes and, more generally, of the tendency in certain research areas to view pervasive positive selection as a foregone conclusion.
基于核苷酸变异模式准确识别和量化与软选择清除相关的遗传特征的能力一直存在争议。我们在这里提供了对最近发表在《PLOS 遗传学》上的一些论文的反驳观点,这些论文不仅认为软选择清除在统计学上是可识别的,而且还认为它们在果蝇和 HIV 群体中普遍具有进化作用。我们提供的证据表明,这些说法归因于缺乏对竞争进化模型的考虑、对经验异常值的不合理解释,以及对这些过程本身的新定义。我们的研究结果突出了在没有适当首先考虑常见过程的情况下,基于假设和偶发过程拟合进化模型的危险,更广泛地说,在某些研究领域存在将普遍的正选择视为既定事实的倾向。