Dhalla Naranjan S, Elimban Vijayan, Bartekova Monika, Adameova Adriana
St. Boniface Hospital Albrechtsen Research Centre, Institute of Cardiovascular Sciences, Department of Physiology and Pathophysiology, Max Rady College of Medicine, University of Manitoba, Winnipeg, MB R2H 2A6, Canada.
Centre of Experimental Medicine, Institute for Heart Research, Slovak Academy of Sciences, Dubravska cesta 9, 84104 Bratislava, Slovakia.
Biomedicines. 2022 Feb 7;10(2):393. doi: 10.3390/biomedicines10020393.
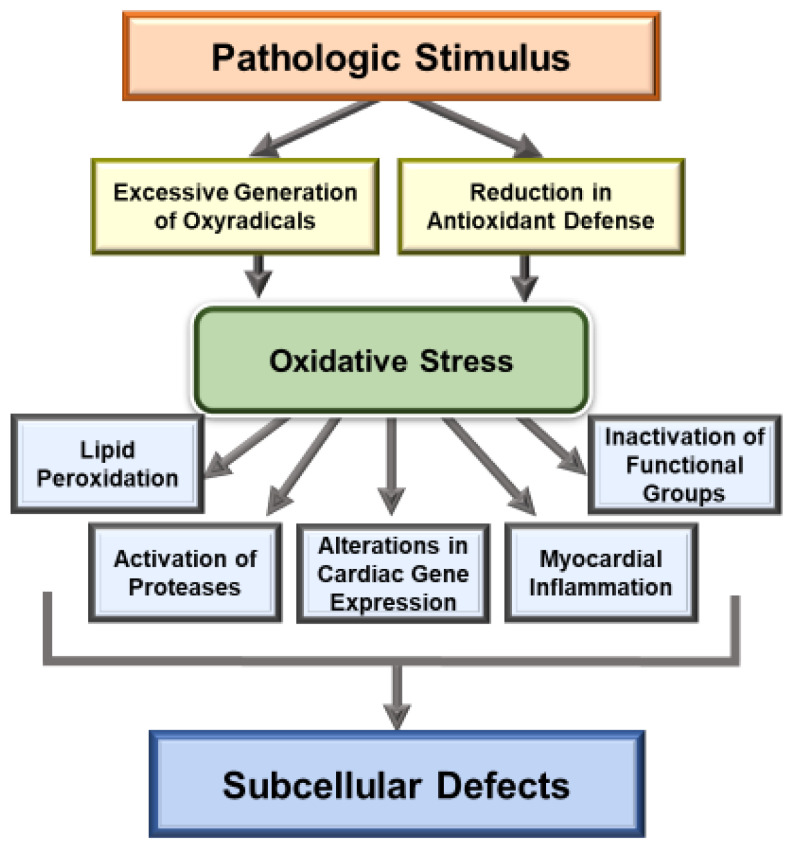
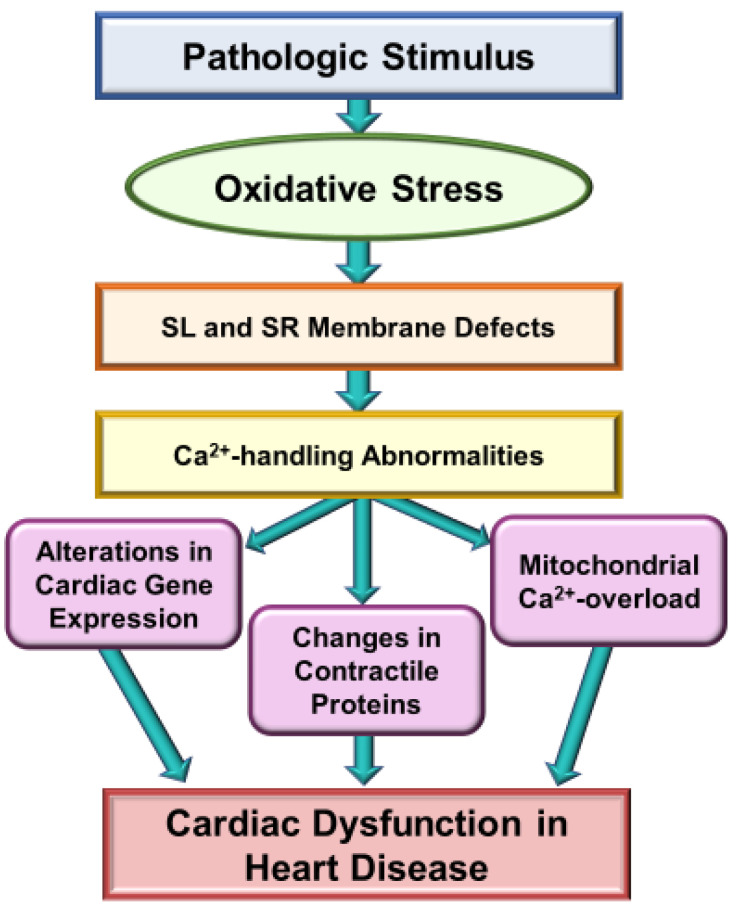
It is now well known that oxidative stress promotes lipid peroxidation, protein oxidation, activation of proteases, fragmentation of DNA and alteration in gene expression for producing myocardial cell damage, whereas its actions for the induction of fibrosis, necrosis and apoptosis are considered to result in the loss of cardiomyocytes in different types of heart disease. The present article is focused on the discussion concerning the generation and implications of oxidative stress from various sources such as defective mitochondrial electron transport and enzymatic reactions mainly due to the activation of NADPH oxidase, nitric oxide synthase and monoamine oxidase in diseased myocardium. Oxidative stress has been reported to promote excessive entry of Ca due to increased permeability of the sarcolemmal membrane as well as depressions of Na-K ATPase and Na-Ca exchange systems, which are considered to increase the intracellular of Ca. In addition, marked changes in the ryanodine receptors and Ca-pump ATPase have been shown to cause Ca-release and depress Ca accumulation in the sarcoplasmic reticulum as a consequence of oxidative stress. Such alterations in sarcolemma and sarcoplasmic reticulum are considered to cause Ca-handling abnormalities, which are associated with mitochondrial Ca-overload and loss of myofibrillar Ca-sensitivity due to oxidative stress. Information regarding the direct effects of different oxyradicals and oxidants on subcellular organelles has also been outlined to show the mechanisms by which oxidative stress may induce Ca-handling abnormalities. These observations support the view that oxidative stress plays an important role in the genesis of subcellular defects and cardiac dysfunction in heart disease.
现在众所周知,氧化应激会促进脂质过氧化、蛋白质氧化、蛋白酶激活、DNA断裂以及基因表达改变,从而导致心肌细胞损伤,而其诱导纤维化、坏死和凋亡的作用被认为会导致不同类型心脏病中心肌细胞的丧失。本文重点讨论了各种来源氧化应激的产生及其影响,这些来源包括患病心肌中线粒体电子传递缺陷和酶促反应,主要是由于NADPH氧化酶、一氧化氮合酶和单胺氧化酶的激活。据报道,氧化应激会由于肌膜通透性增加以及Na-K ATP酶和Na-Ca交换系统功能降低而促进钙离子过度内流,这被认为会增加细胞内钙离子浓度。此外,已表明兰尼碱受体和钙泵ATP酶的显著变化会导致钙离子释放,并由于氧化应激而降低肌浆网中钙离子的蓄积。肌膜和肌浆网的这种改变被认为会导致钙离子处理异常,这与氧化应激引起的线粒体钙超载和肌原纤维钙敏感性丧失有关。本文还概述了不同氧自由基和氧化剂对亚细胞器的直接影响,以说明氧化应激可能诱导钙离子处理异常的机制。这些观察结果支持了氧化应激在心脏病亚细胞缺陷和心脏功能障碍发生中起重要作用的观点。