Wang NingJie, Chen ShuiFei, Xie Lei, Wang Lu, Feng YueYao, Lv Ting, Fang YanMing, Ding Hui
Co-Innovation Center for Sustainable Forestry in Southern China College of Biology and the Environment Key Laboratory of State Forestry and Grassland Administration on Subtropical Forest Biodiversity Conservation Nanjing Forestry University Nanjing China.
Research Center for Nature Conservation and Biodiversity State Environmental Protection Scientific Observation and Research Station for Ecology and Environment of Wuyi Mountains State Environmental Protection Key Laboratory on Biosafety Nanjing Institute of Environmental Sciences, Ministry of Ecology and Environment Nanjing China.
Ecol Evol. 2022 Feb 16;12(2):e8637. doi: 10.1002/ece3.8637. eCollection 2022 Feb.
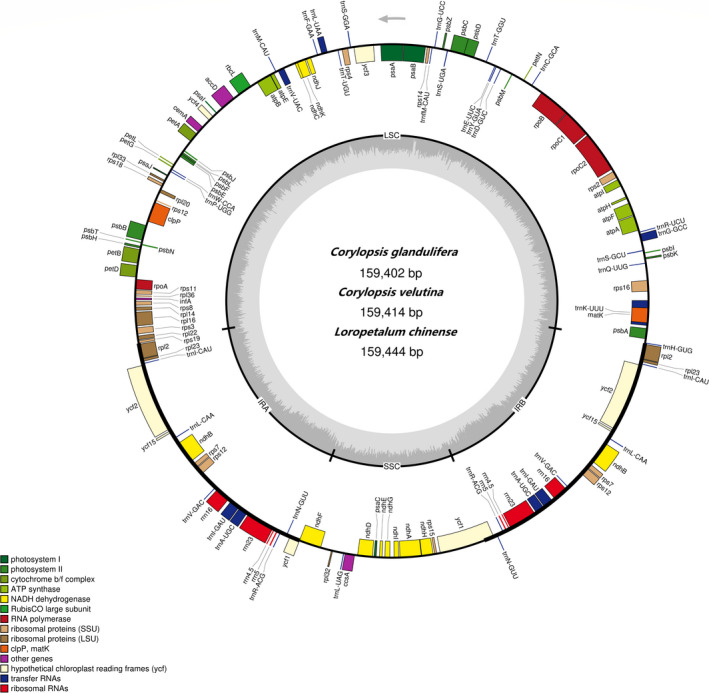
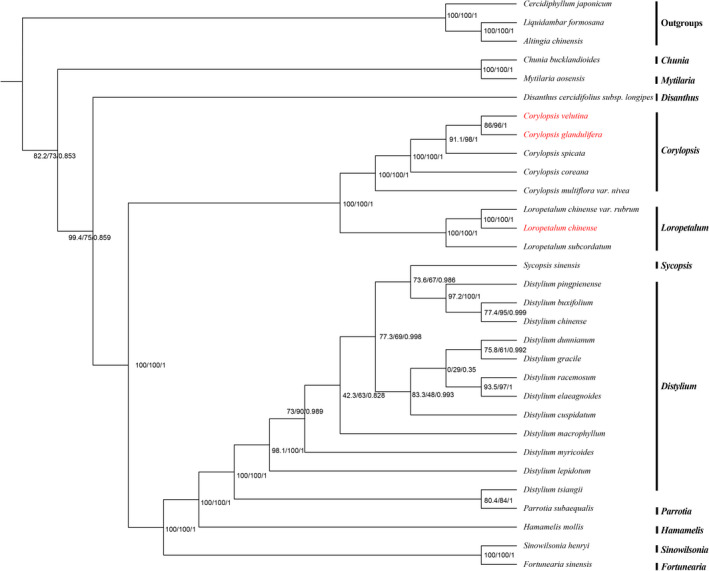
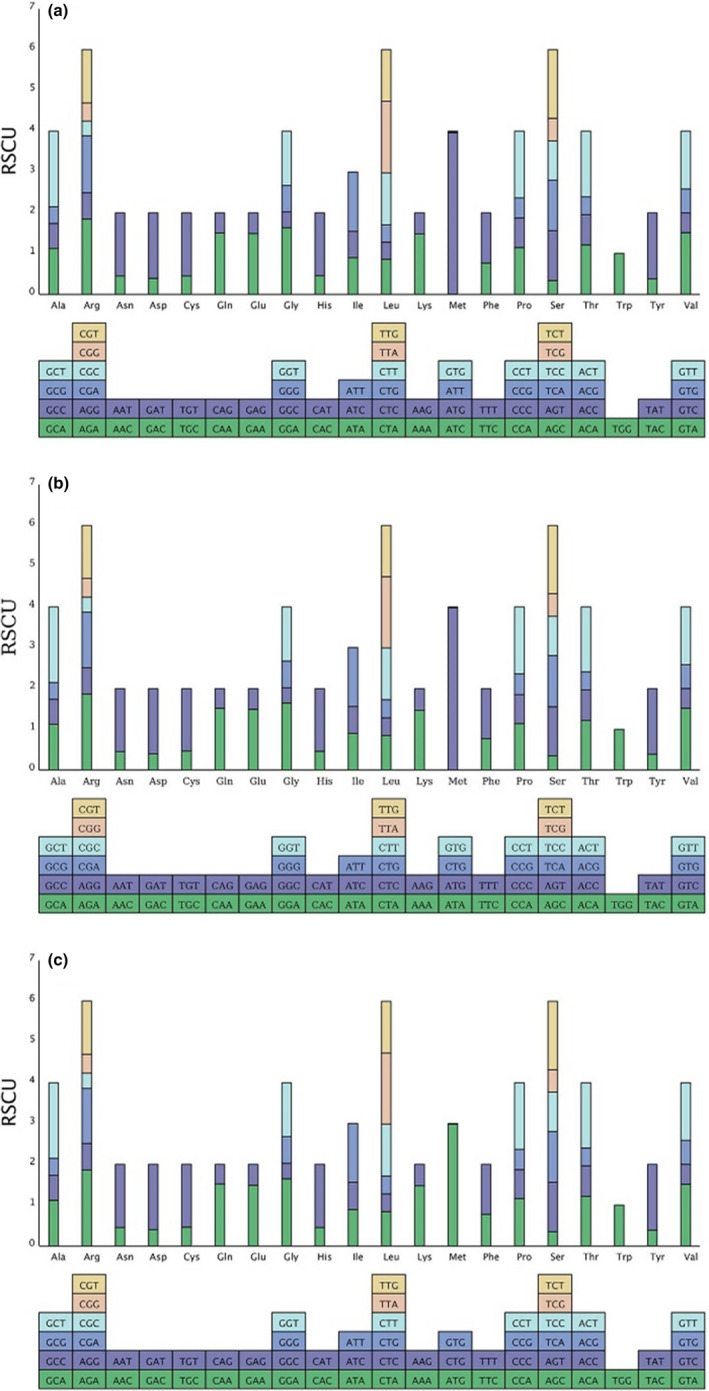
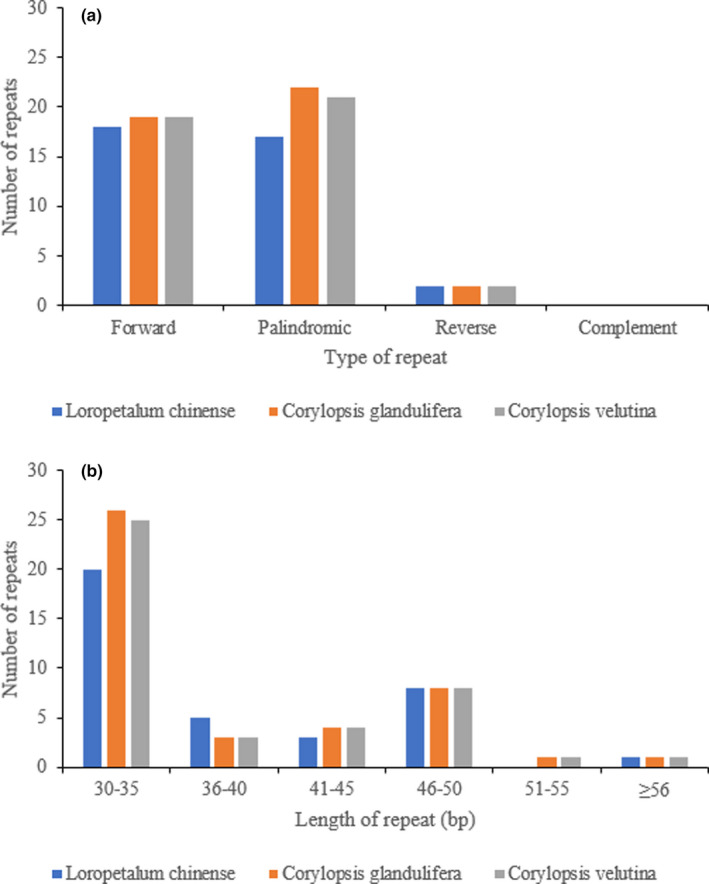
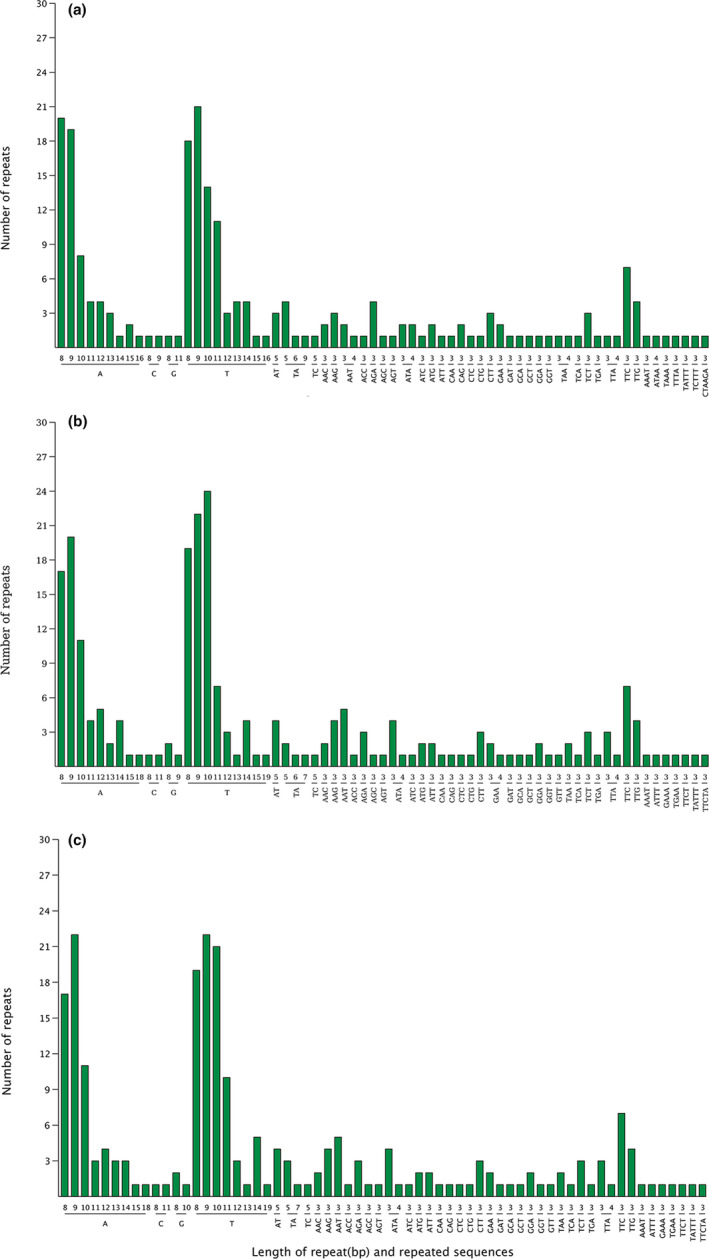
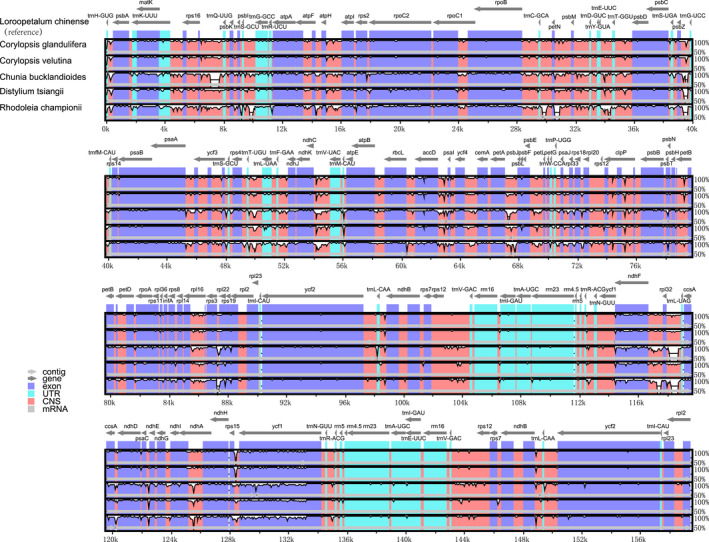
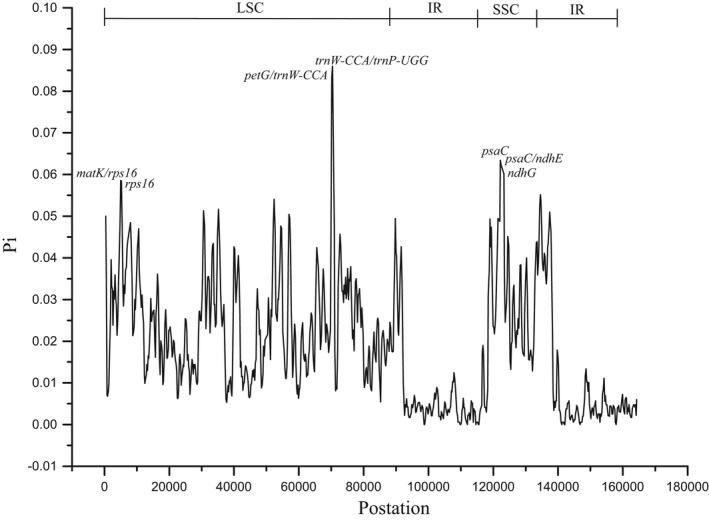
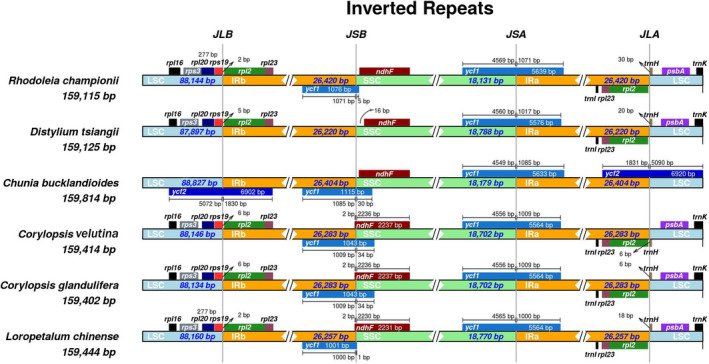
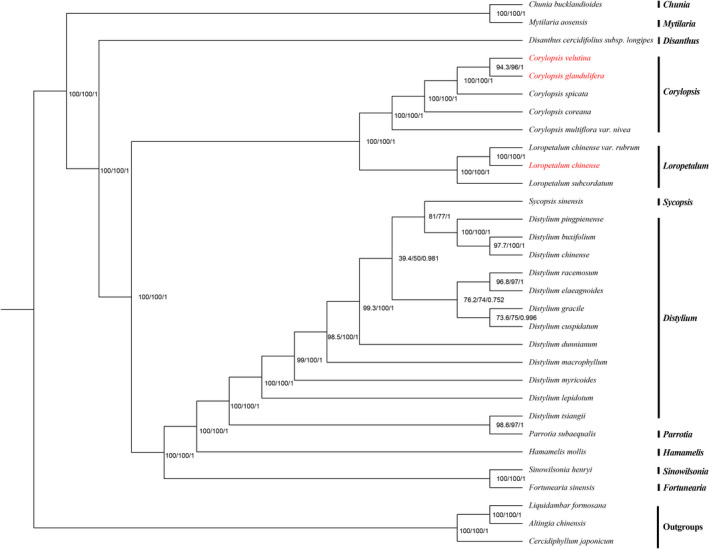
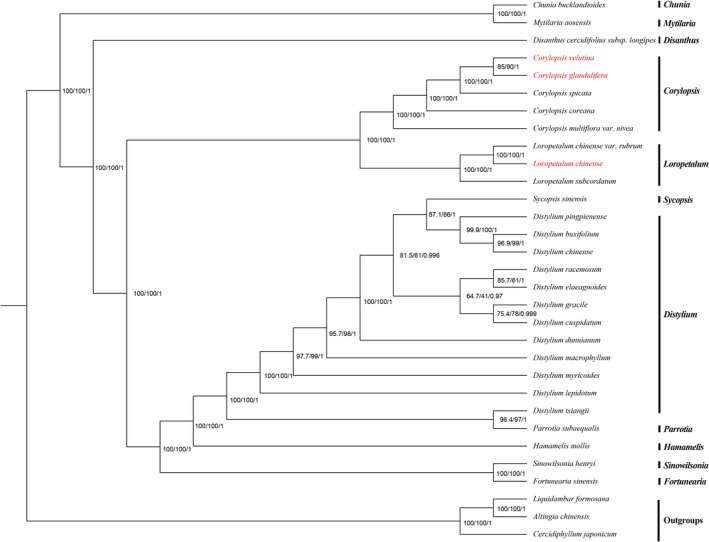
Hamamelidaceae is an important group that represents the origin and early evolution of angiosperms. Its plants have many uses, such as timber, medical, spice, and ornamental uses. In this study, the complete chloroplast genomes of (R. Br.) Oliver, Hemsl., and Hand.-Mazz. were sequenced using the Illumina NovaSeq 6000 platform. The sizes of the three chloroplast genomes were 159,402 bp (. ), 159,414 bp (. ), and 159,444 bp (. ), respectively. These chloroplast genomes contained typical quadripartite structures with a pair of inverted repeat (IR) regions (26,283, 26,283, and 26,257 bp), a large single-copy (LSC) region (88,134, 88,146, and 88,160 bp), and a small single-copy (SSC) region (18,702, 18,702, and 18,770 bp). The chloroplast genomes encoded 132-133 genes, including 85-87 protein-coding genes, 37-38 tRNA genes, and 8 rRNA genes. The coding regions were composed of 26,797, 26,574, and 26,415 codons, respectively, most of which ended in A/U. A total of 37-43 long repeats and 175-178 simple sequence repeats (SSRs) were identified, and the SSRs contained a higher number of A + T than G + C bases. The genome comparison showed that the IR regions were more conserved than the LSC or SSC regions, while the noncoding regions contained higher variability than the gene coding regions. Phylogenetic analyses revealed that species in the same genus tended to cluster together. Hung T. Chang, Lecomte, and Maxim. may have diverged early and Siebold & Zucc. was closely related to R. Br. This study provides valuable information for further species identification, evolution, and phylogenetic studies of Hamamelidaceae plants.
金缕梅科是一个重要的类群,代表了被子植物的起源和早期进化。其植物有许多用途,如木材、医药、香料和观赏用途。在本研究中,使用Illumina NovaSeq 6000平台对(R. Br.)Oliver、Hemsl.和Hand.-Mazz.的完整叶绿体基因组进行了测序。这三个叶绿体基因组的大小分别为159,402 bp(. )、159,414 bp(. )和159,444 bp(. )。这些叶绿体基因组包含典型的四分体结构,有一对反向重复(IR)区域(26,283、26,283和26,257 bp)、一个大单拷贝(LSC)区域(88,134、88,146和88,160 bp)和一个小单拷贝(SSC)区域(18,702、18,702和18,770 bp)。叶绿体基因组编码132 - 133个基因,包括85 - 87个蛋白质编码基因、37 - 38个tRNA基因和8个rRNA基因。编码区分别由26,797、26,574和26,415个密码子组成,其中大多数以A/U结尾。共鉴定出37 - 43个长重复序列和175 - 178个简单序列重复(SSR),且SSR中A + T碱基的数量高于G + C碱基。基因组比较表明,IR区域比LSC或SSC区域更保守,而非编码区域的变异性高于基因编码区域。系统发育分析表明,同一属的物种倾向于聚类在一起。Hung T. Chang、Lecomte和Maxim.可能分歧较早,而Siebold & Zucc.与R. Br.关系密切。本研究为金缕梅科植物的进一步物种鉴定、进化和系统发育研究提供了有价值的信息。