Deakin University, School of Life and Environmental Sciences, Geelong, Australia.
College of Health and Medical Technology, Middle Technical University, Baghdad, Iraq.
PLoS Comput Biol. 2022 Mar 9;18(3):e1009935. doi: 10.1371/journal.pcbi.1009935. eCollection 2022 Mar.
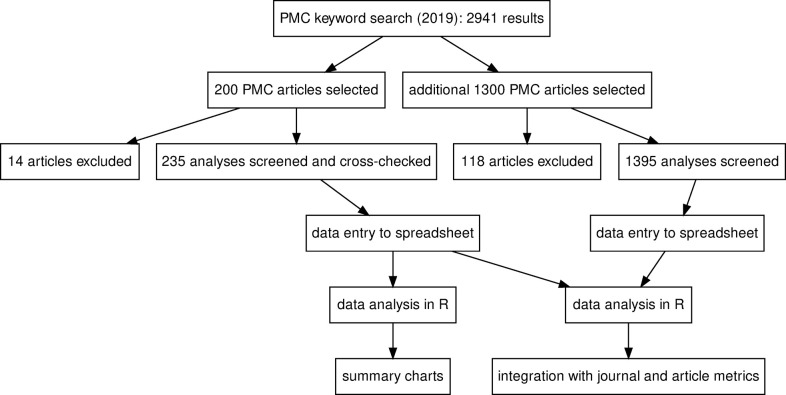
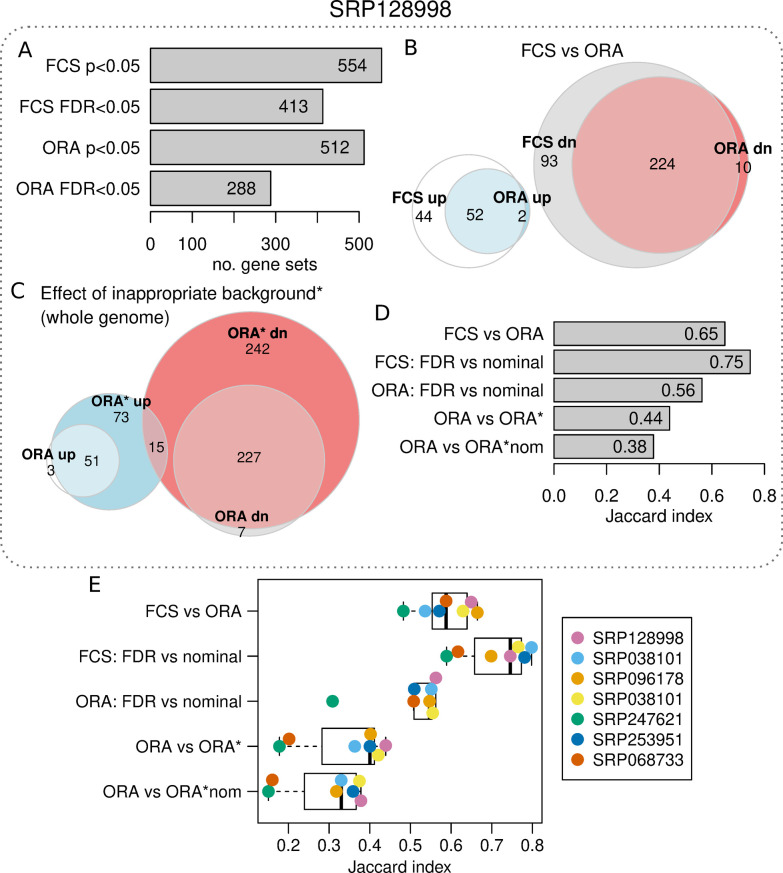
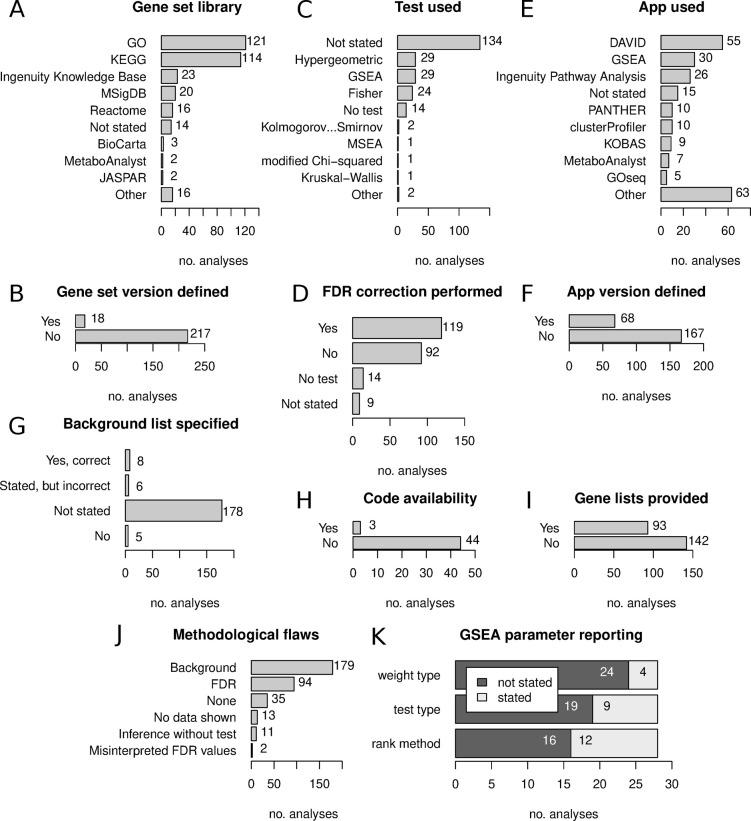
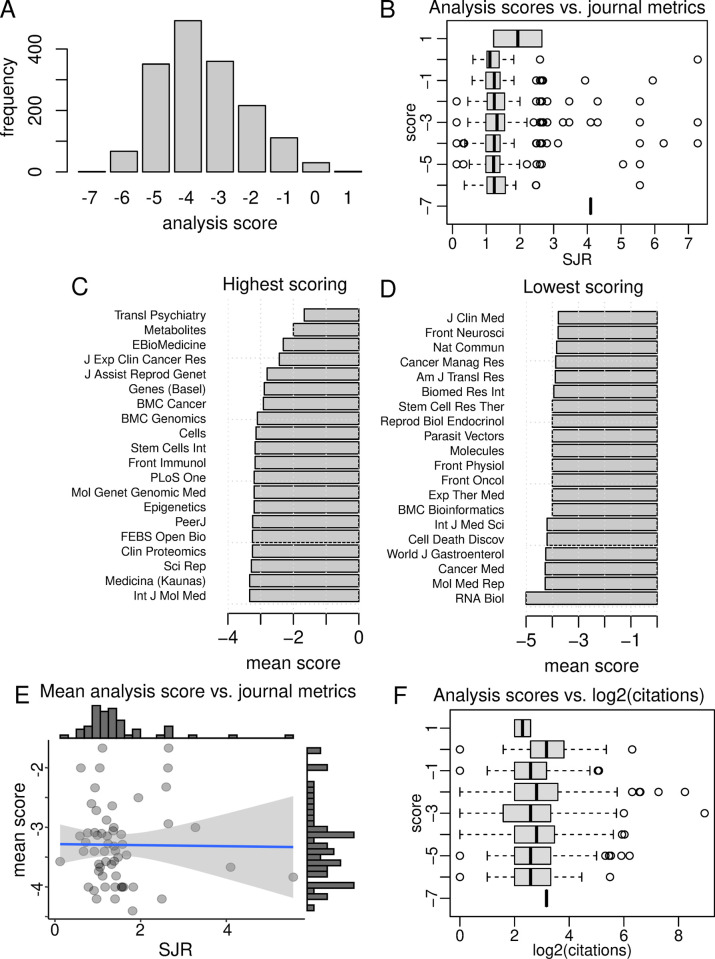
Gene set enrichment tests (a.k.a. functional enrichment analysis) are among the most frequently used methods in computational biology. Despite this popularity, there are concerns that these methods are being applied incorrectly and the results of some peer-reviewed publications are unreliable. These problems include the use of inappropriate background gene lists, lack of false discovery rate correction and lack of methodological detail. To ascertain the frequency of these issues in the literature, we performed a screen of 186 open-access research articles describing functional enrichment results. We find that 95% of analyses using over-representation tests did not implement an appropriate background gene list or did not describe this in the methods. Failure to perform p-value correction for multiple tests was identified in 43% of analyses. Many studies lacked detail in the methods section about the tools and gene sets used. An extension of this survey showed that these problems are not associated with journal or article level bibliometrics. Using seven independent RNA-seq datasets, we show misuse of enrichment tools alters results substantially. In conclusion, most published functional enrichment studies suffered from one or more major flaws, highlighting the need for stronger standards for enrichment analysis.
基因集富集测试(也称为功能富集分析)是计算生物学中最常用的方法之一。尽管如此,人们担心这些方法的应用不正确,一些经过同行评审的出版物的结果不可靠。这些问题包括使用不适当的背景基因列表、缺乏错误发现率校正以及缺乏方法学细节。为了确定这些问题在文献中的频率,我们对 186 篇描述功能富集结果的开放获取研究文章进行了筛选。我们发现,95%使用过度表达测试的分析没有使用适当的背景基因列表,或者在方法中没有描述这一点。在 43%的分析中,没有对多个测试进行 p 值校正。许多研究在方法部分缺乏关于所使用的工具和基因集的详细信息。这项调查的扩展表明,这些问题与期刊或文章级别的文献计量学无关。使用七个独立的 RNA-seq 数据集,我们表明富集工具的误用会大大改变结果。总之,大多数已发表的功能富集研究都存在一个或多个主要缺陷,这突出表明需要对富集分析制定更强的标准。