Yang Chao, Wang Zeng, Hu Yuanchang, Yang Shikun, Cheng Feng, Rao Jianhua, Wang Xuehao
Hepatobiliary Center, The First Affiliated Hospital of Nanjing Medical University; Key Laboratory of Liver Transplantation, Chinese Academy of Medical Sciences, 210029, Nanjing, Jiangsu Province, China.
Cell Death Discov. 2022 Mar 14;8(1):115. doi: 10.1038/s41420-022-00910-z.
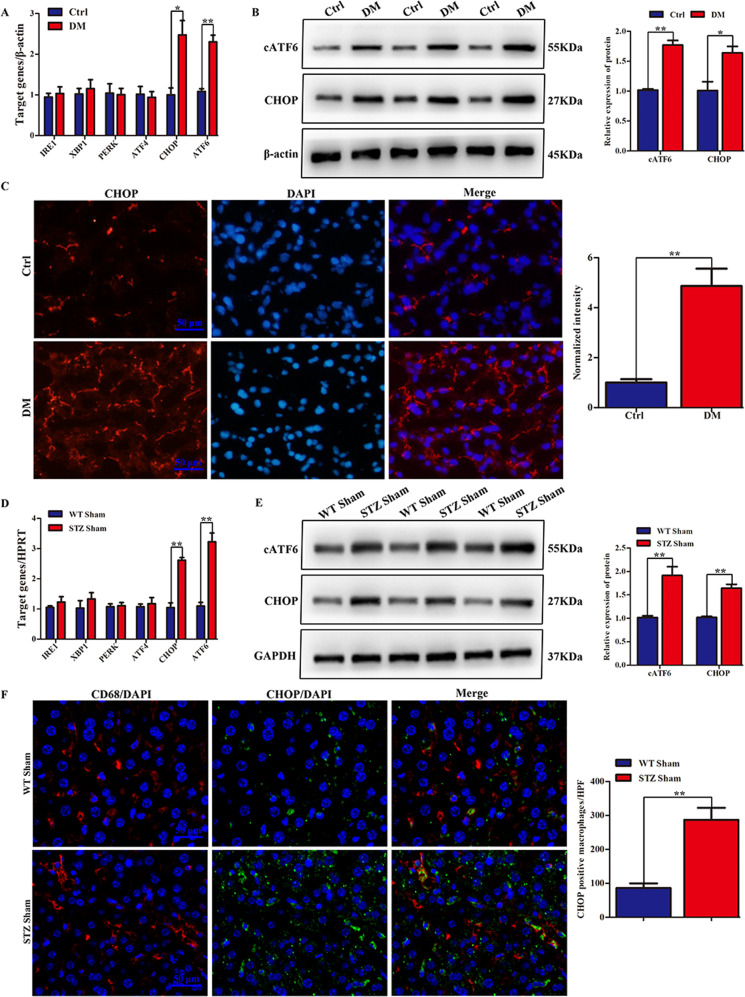
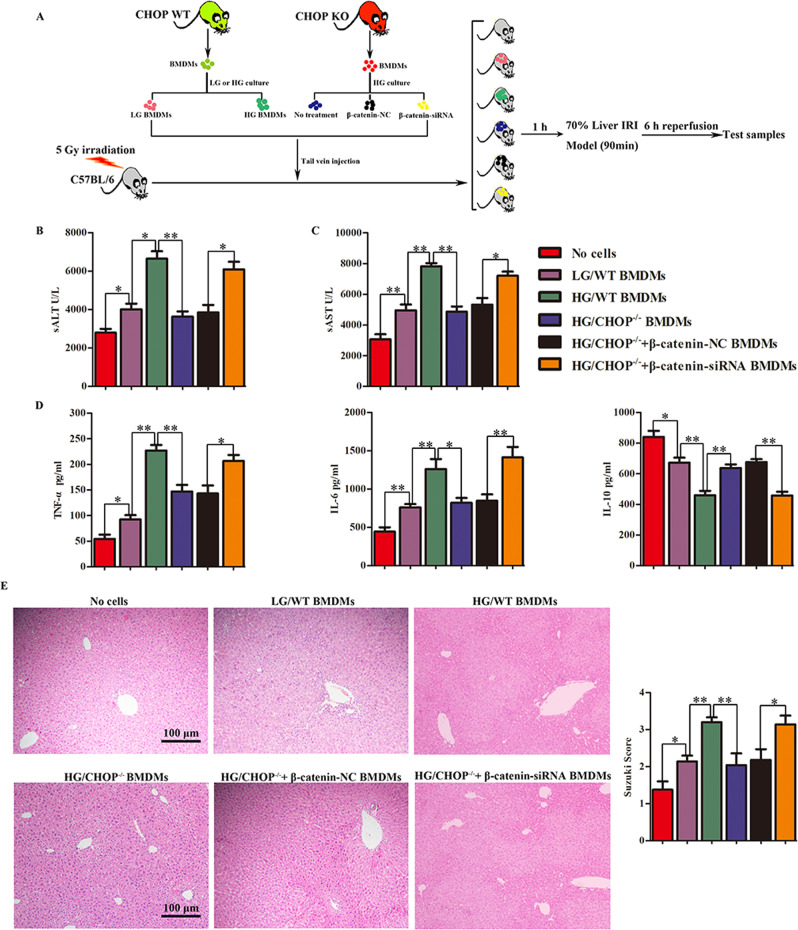
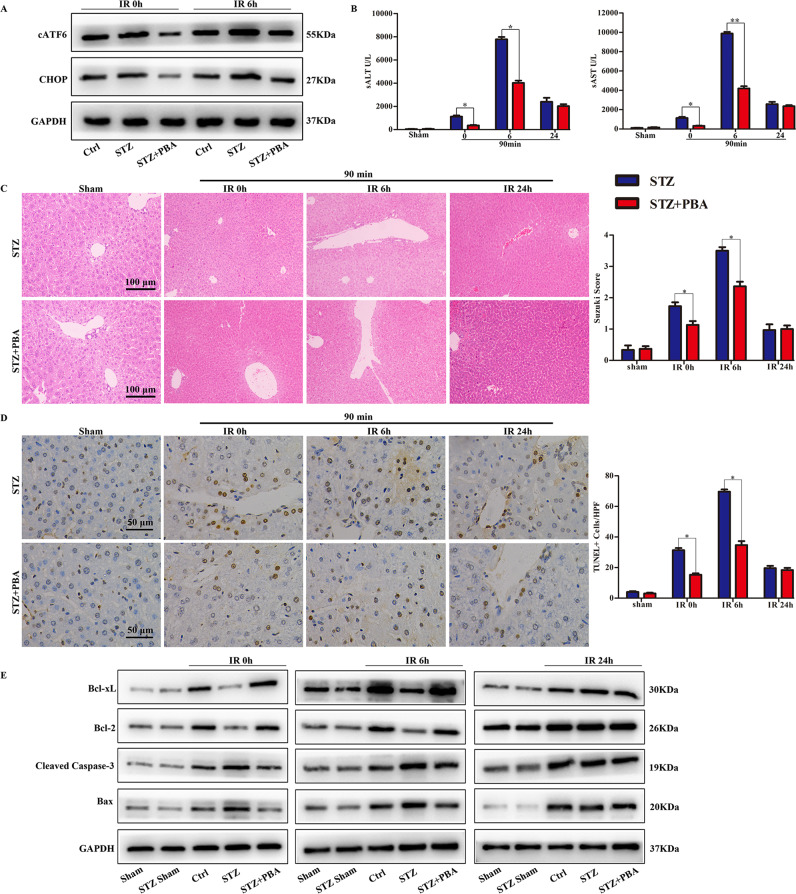
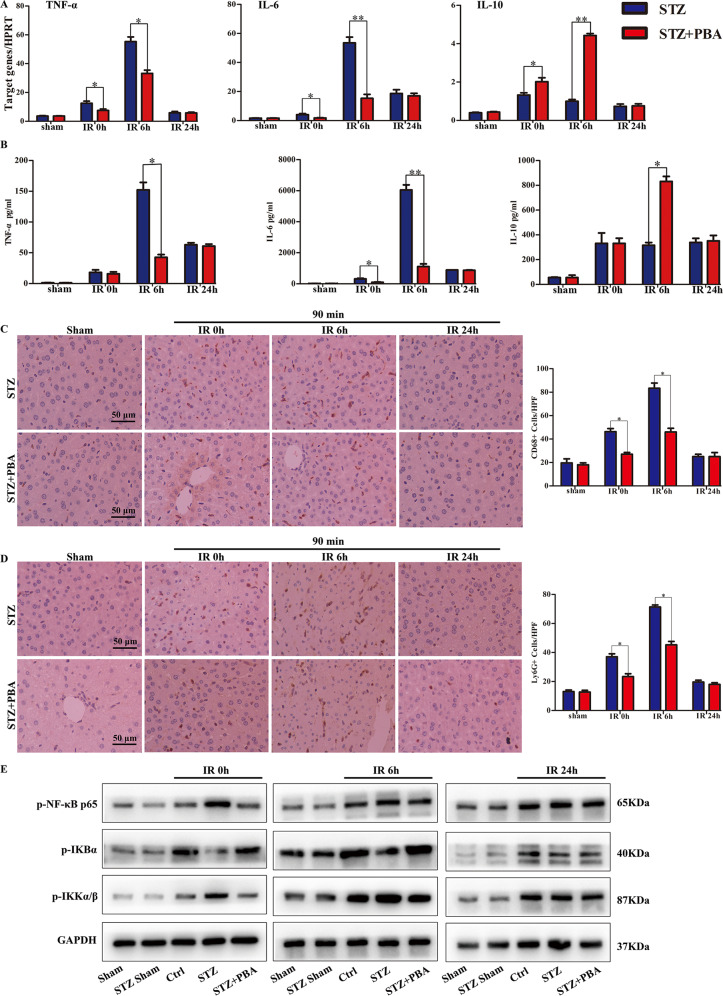
Although hyperglycemia has been documented as an unfavorable element that can further induce liver ischemia-reperfusion injury (IRI), the related molecular mechanisms remain to be clearly elaborated. This study investigated the effective manner of endoplasmic reticulum (ER) stress signaling in hyperglycemia-exacerbated liver IRI. Here we demonstrated that in the liver tissues and Kupffer cells (KCs) of DM patients and STZ-induced hyperglycemic mice, the ER stress-ATF6-CHOP signaling pathway is activated. TLR4-mediated pro-inflammatory activation was greatly attenuated by the addition of 4-phenylbutyrate (PBA), one common ER stress inhibitor. The liver IRI in hyperglycemic mice was also significantly reduced after PBA treatment. In addition, deficiency of CHOP (CHOP) obviously alleviates the hepatic IRI, and pro-inflammatory effects deteriorated by hyperglycemia. In hyperglycemic mice, β-catenin expression was suppressed while the ATF6-CHOP signal was activated. In the liver tissues of PBA-treated or CHOP hyperglycemic mice, the expression of β-catenin was restored. Furthermore, CHOP deficiency can induce protection against hyperglycemia-related liver IRI, which was disrupted by the knockdown of β-catenin will cause this protection to disappear. High glucose (HG) treatment stimulated ATF6-CHOP signaling, reduced cellular β-catenin accumulation, and promoted the TLR4-related inflammation of BMDMs. But the above effects were partially rescued in BMDMs with CHOP deficiency or by PBA treatment. In BMDMs cultured in HG conditions, the anti-inflammatory functions of CHOP were destroyed by the knockdown of β-catenin. Finally, chimeric mice carrying WT or CHOP BMDMs by bone marrow transplantation were adopted to verify the above conclusion. The current study suggested that hyperglycemia could trigger ER stress-ATF6-CHOP axis, inhibit β-catenin activation, accelerate inflammation, and deteriorate liver IRI, thus providing the treatment potential for management of sterile liver inflammation in DM patients.
尽管高血糖已被证明是一种不利因素,可进一步诱发肝脏缺血再灌注损伤(IRI),但其相关分子机制仍有待明确阐述。本研究探讨了内质网(ER)应激信号在高血糖加重的肝脏IRI中的作用机制。我们发现,在糖尿病患者及链脲佐菌素诱导的高血糖小鼠的肝脏组织和库普弗细胞(KCs)中,ER应激-ATF6-CHOP信号通路被激活。添加一种常见的ER应激抑制剂4-苯基丁酸盐(PBA)可显著减弱TLR4介导的促炎激活。PBA处理后,高血糖小鼠的肝脏IRI也显著降低。此外,CHOP基因敲除(CHOP)明显减轻了肝脏IRI以及高血糖恶化的促炎作用。在高血糖小鼠中,β-连环蛋白表达受到抑制,而ATF6-CHOP信号被激活。在PBA处理的高血糖小鼠或CHOP基因敲除的高血糖小鼠的肝脏组织中,β-连环蛋白的表达得以恢复。此外,CHOP基因敲除可诱导对高血糖相关肝脏IRI的保护作用,而β-连环蛋白的敲低会破坏这种保护作用。高糖(HG)处理可刺激ATF6-CHOP信号,减少细胞内β-连环蛋白的积累,并促进骨髓来源的巨噬细胞(BMDMs)的TLR4相关炎症反应。但在CHOP基因敲除的BMDMs或PBA处理的BMDMs中,上述作用部分得到挽救。在HG条件下培养的BMDMs中,β-连环蛋白的敲低破坏了CHOP的抗炎功能。最后,通过骨髓移植构建携带野生型或CHOP基因敲除BMDMs的嵌合小鼠,验证上述结论。本研究表明,高血糖可触发ER应激-ATF6-CHOP轴,抑制β-连环蛋白激活,加速炎症反应,加重肝脏IRI,从而为糖尿病患者无菌性肝脏炎症的治疗提供了潜在靶点。