Cancer Genetics Laboratory, Kolling Institute, Royal North Shore Hospital, Sydney, NSW, 2065, Australia.
The University of Sydney, Sydney, NSW, 2006, Australia.
J Clin Endocrinol Metab. 2022 Jul 14;107(8):2339-2349. doi: 10.1210/clinem/dgac162.
Germline CDKN1B pathogenic variants result in multiple endocrine neoplasia type 4 (MEN4), an autosomal dominant hereditary tumor syndrome variably associated with primary hyperparathyroidism, pituitary adenoma, and duodenopancreatic neuroendocrine tumors.
To report the phenotype of 3 unrelated cases each with a unique germline CDKN1B variant (of which 2 are novel) and compare these cases with those described in the current literature.
DESIGN/METHODS: Three case studies, including clinical presentation, germline, and tumor genetic analysis and family history.
Two tertiary University Hospitals in Sydney, New South Wales, and 1 tertiary University Hospital in Canberra, Australian Capital Territory, Australia.
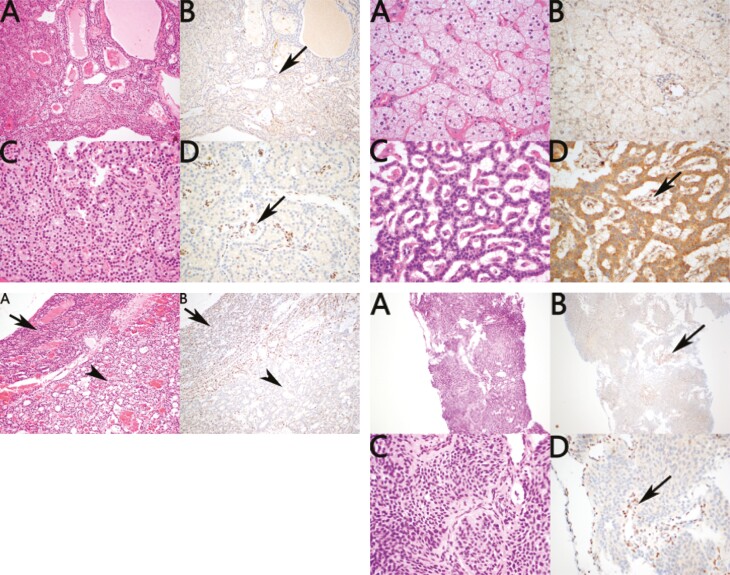
Phenotype of the 3 cases and their kindred; molecular analysis and tumor p27kip1 immunohistochemistry.
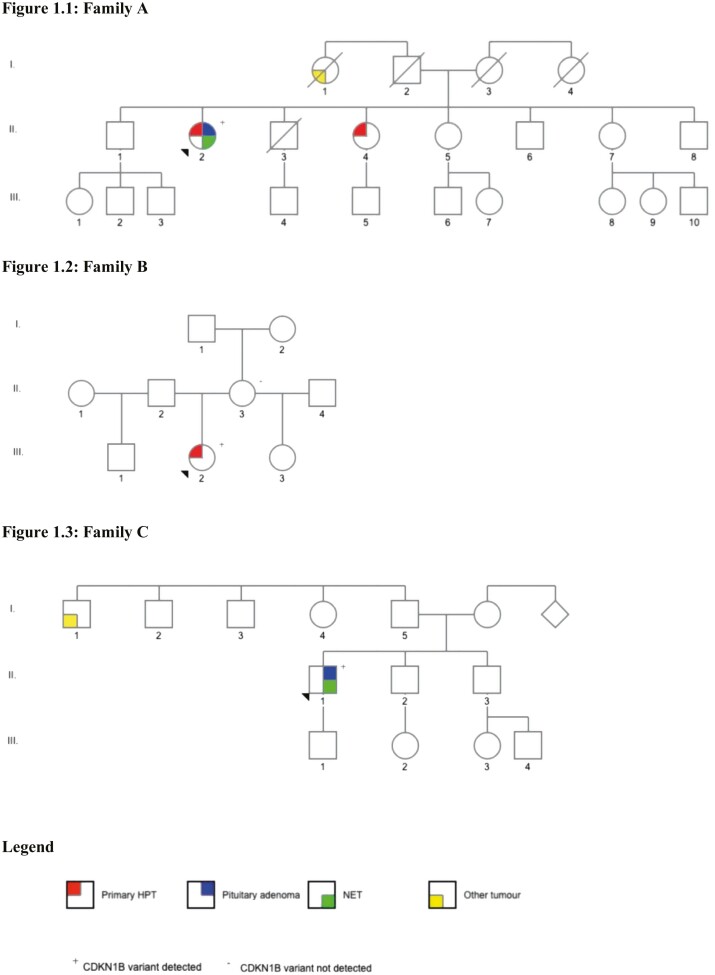
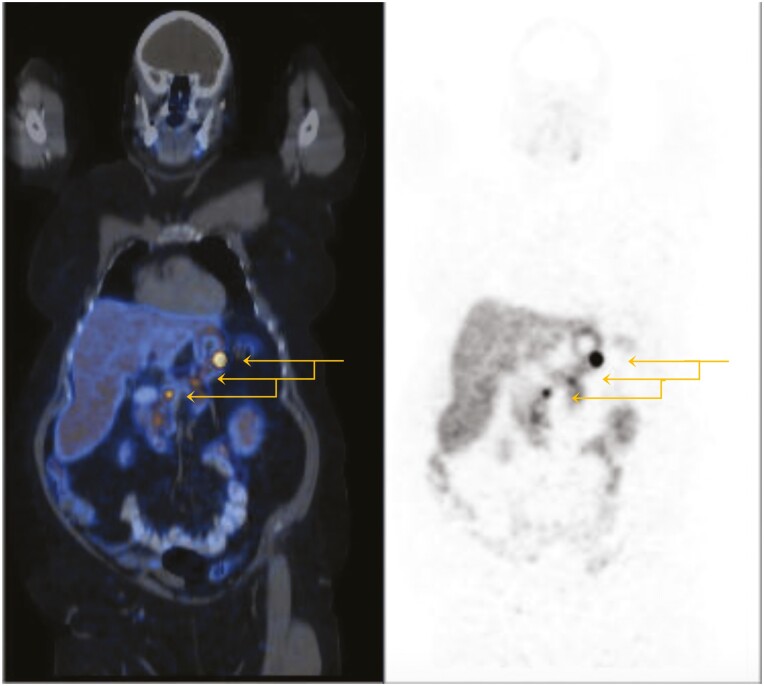
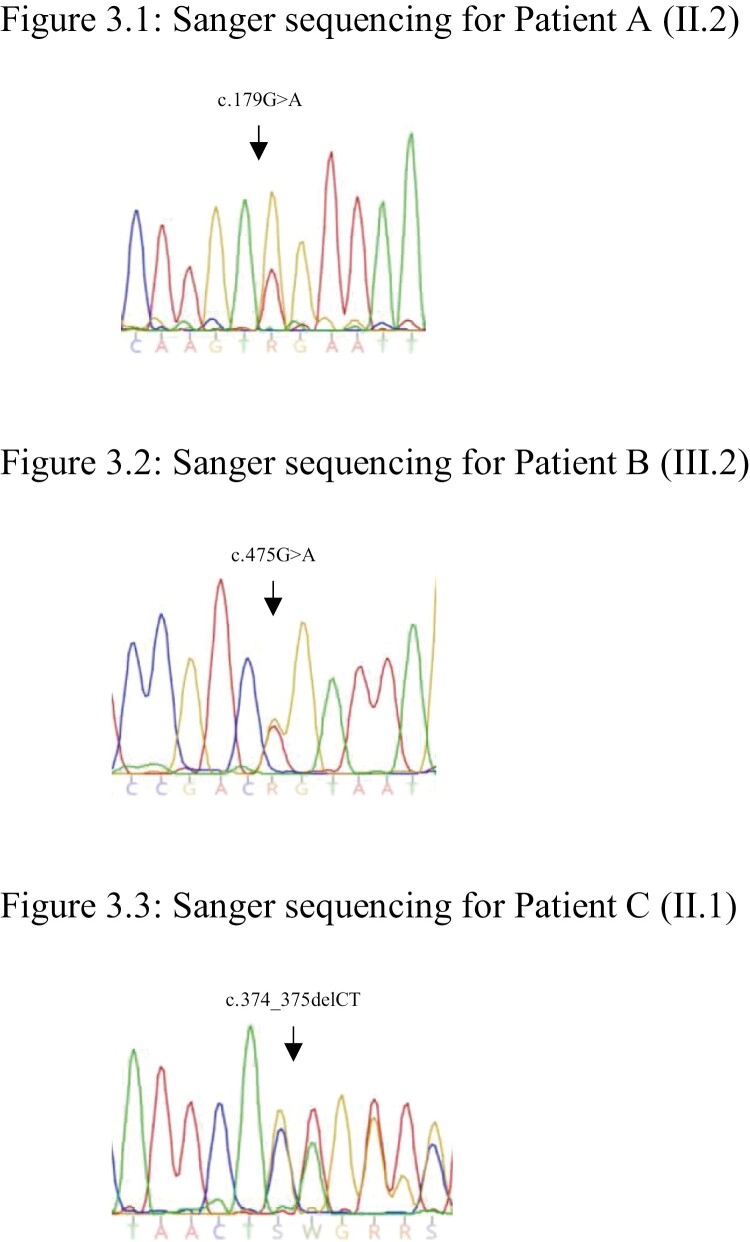
Family A: The proband developed multiglandular primary hyperparathyroidism, a microprolactinoma and a multifocal nonfunctioning duodenopancreatic neuroendocrine tumor. Family B: The proband was diagnosed with primary hyperparathyroidism from a single parathyroid adenoma. Family C: The proband was diagnosed with a nonfunctioning pituitary microadenoma and ectopic Cushing's syndrome from an atypical thymic carcinoid tumor. Germline sequencing in each patient identified a unique variant in CDKN1B, 2 of which are novel (c.179G > A, p.Trp60*; c.475G > A, p.Asp159Asn) and 1 previously reported (c.374_375delCT, p.Ser125*).
Germline CDKN1B pathogenic variants cause the syndrome MEN4. The phenotype resulting from the 3 pathogenic variants described in this series highlights the heterogenous nature of this syndrome, ranging from isolated primary hyperparathyroidism to the full spectrum of endocrine manifestations. We report the first described cases of a prolactinoma and an atypical thymic carcinoid tumor in MEN4.
胚系 CDKN1B 致病性变异可导致多发性内分泌肿瘤 4 型(MEN4),这是一种常染色体显性遗传性肿瘤综合征,与甲状旁腺功能亢进、垂体腺瘤和十二指肠胰腺神经内分泌肿瘤有关。
报告 3 例具有独特胚系 CDKN1B 变异(其中 2 例为新变异)的散发性病例,并与目前文献中的病例进行比较。
设计/方法:3 例病例研究,包括临床表现、胚系和肿瘤遗传分析以及家族史。
澳大利亚新南威尔士州悉尼的 2 家三级大学医院和澳大利亚首都领地堪培拉的 1 家三级大学医院。
3 例病例及其亲属的表型;分子分析和肿瘤 p27kip1 免疫组化。
家族 A:先证者患有多腺体原发性甲状旁腺功能亢进症、微催乳素瘤和多灶性无功能性十二指肠胰腺神经内分泌肿瘤。家族 B:先证者因单个甲状旁腺腺瘤而被诊断为原发性甲状旁腺功能亢进症。家族 C:先证者因非功能性垂体微腺瘤和异位库欣综合征而被诊断为非典型胸腺癌。每位患者的胚系测序均发现 CDKN1B 中的一个独特变异,其中 2 个为新变异(c.179G>A,p.Trp60*;c.475G>A,p.Asp159Asn),1 个为先前报道的变异(c.374_375delCT,p.Ser125*)。
胚系 CDKN1B 致病性变异导致 MEN4 综合征。本系列中描述的 3 个致病性变异导致的表型突出了该综合征的异质性,从孤立性原发性甲状旁腺功能亢进症到所有内分泌表现均有涉及。我们报告了 MEN4 中首例描述的催乳素瘤和非典型胸腺癌病例。