St. Petersburg State University, 199034 St. Petersburg, Russia.
City Hospital No. 40, 197706 St. Petersburg, Russia.
Genes (Basel). 2022 Mar 17;13(3):534. doi: 10.3390/genes13030534.
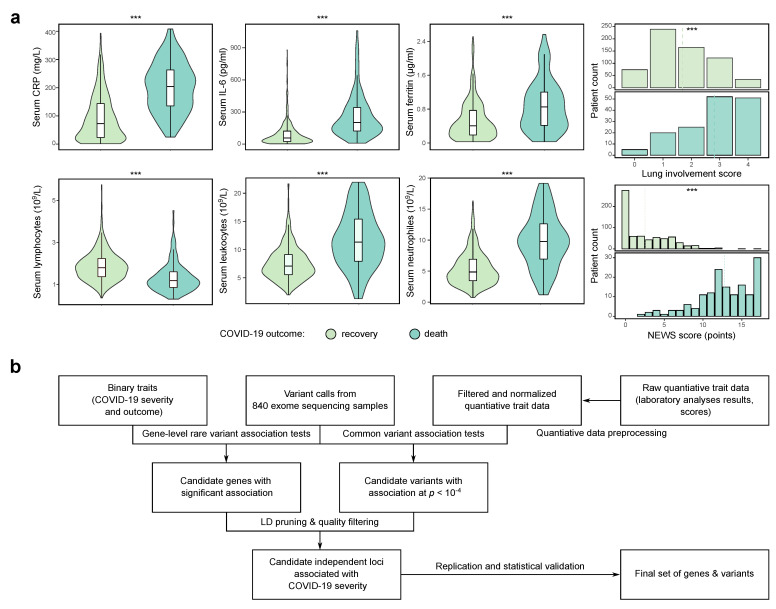
The COVID-19 pandemic has drawn the attention of many researchers to the interaction between pathogen and host genomes. Over the last two years, numerous studies have been conducted to identify the genetic risk factors that predict COVID-19 severity and outcome. However, such an analysis might be complicated in cohorts of limited size and/or in case of limited breadth of genome coverage. In this work, we tried to circumvent these challenges by searching for candidate genes and genetic variants associated with a variety of quantitative and binary traits in a cohort of 840 COVID-19 patients from Russia. While we found no gene- or pathway-level associations with the disease severity and outcome, we discovered eleven independent candidate loci associated with quantitative traits in COVID-19 patients. Out of these, the most significant associations correspond to rs1651553 in = 1.4 × 10), rs11243705 in ( = 8.2 × 10), and rs16885 in ( = 1.3 × 10). One of the identified variants, rs33985936 in , was successfully replicated in an independent study, and three of the variants were found to be associated with blood-related quantitative traits according to the UK Biobank data (rs33985936 in , rs16885 in , and rs4747194 in ). Moreover, we show that a risk score based on these variants can predict the severity and outcome of hospitalization in our cohort of patients. Given these findings, we believe that our work may serve as proof-of-concept study demonstrating the utility of quantitative traits and extensive phenotyping for identification of genetic risk factors of severe COVID-19.
COVID-19 大流行引起了许多研究人员对病原体和宿主基因组相互作用的关注。在过去的两年中,已经进行了许多研究来确定预测 COVID-19 严重程度和结果的遗传风险因素。然而,在规模有限的队列中或在基因组覆盖范围有限的情况下,这种分析可能会很复杂。在这项工作中,我们试图通过在来自俄罗斯的 840 名 COVID-19 患者队列中搜索与各种定量和二进制特征相关的候选基因和遗传变异来规避这些挑战。虽然我们没有发现与疾病严重程度和结果相关的基因或途径水平的关联,但我们发现了 11 个与 COVID-19 患者定量特征相关的独立候选基因座。其中,最显著的关联对应于 rs1651553 在 ( = 1.4 × 10)、rs11243705 在 ( = 8.2 × 10)和 rs16885 在 ( = 1.3 × 10)。鉴定出的变异体之一,即 rs33985936 在 中,在独立研究中成功复制,并且根据 UK Biobank 数据发现三个变异体与血液相关的定量特征相关(rs33985936 在 中,rs16885 在 中,和 rs4747194 在 中)。此外,我们表明,基于这些变体的风险评分可以预测我们患者队列中住院严重程度和结果。鉴于这些发现,我们认为我们的工作可以作为概念验证研究,证明定量特征和广泛表型在确定严重 COVID-19 的遗传风险因素方面的效用。