Allen Institute for Immunology, Seattle, WA, USA.
Department of Genome Sciences, University of Washington School of Medicine, Seattle, WA, USA.
BMC Bioinformatics. 2022 Mar 27;23(1):106. doi: 10.1186/s12859-022-04620-2.
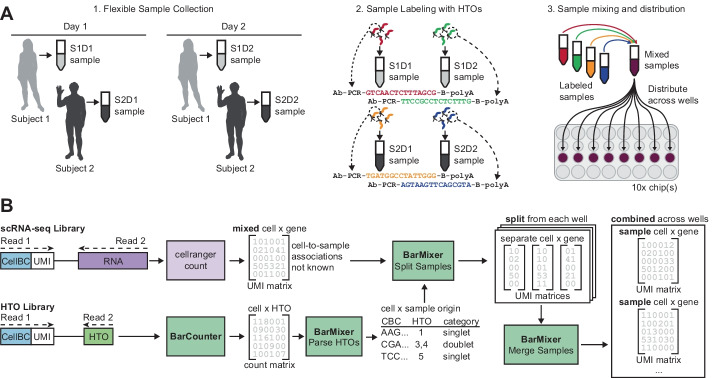
Barcode-based multiplexing methods can be used to increase throughput and reduce batch effects in large single-cell genomics studies. Despite advantages in flexibility of sample collection and scale, there are additional complications in the data deconvolution steps required to assign each cell to their originating samples.
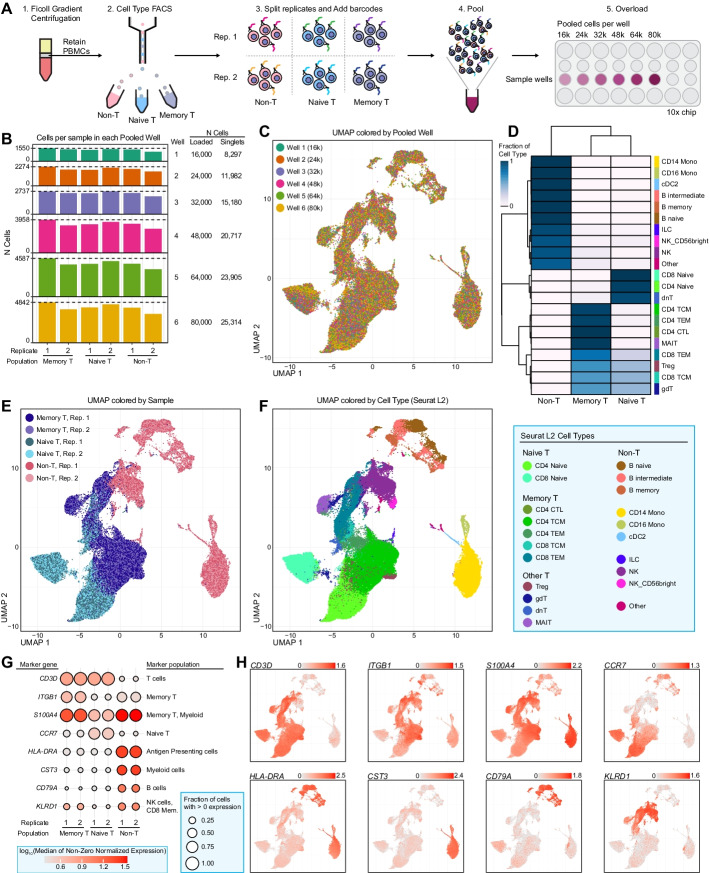
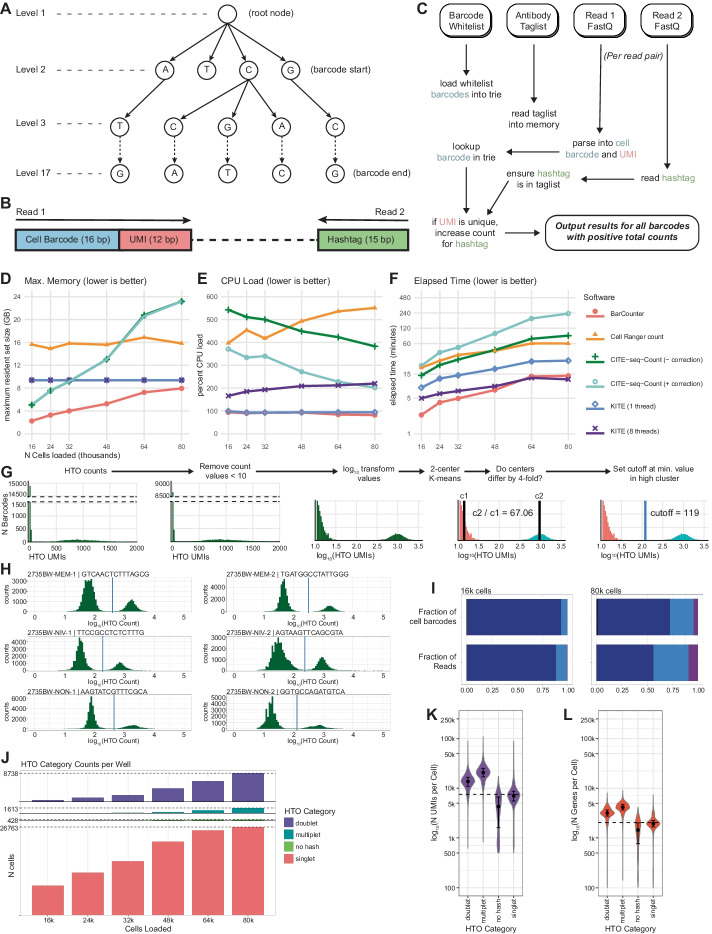
To meet computational needs for efficient sample deconvolution, we developed the tools BarCounter and BarMixer that compute barcode counts and deconvolute mixed single-cell data into sample-specific files, respectively. Together, these tools are implemented as the BarWare pipeline to support demultiplexing from large sequencing projects with many wells of hashed 10x Genomics scRNA-seq data.
BarWare is a modular set of tools linked by shell scripting: BarCounter, a computationally efficient barcode sequence quantification tool implemented in C; and BarMixer, an R package for identification of barcoded populations, merging barcoded data from multiple wells, and quality-control reporting related to scRNA-seq data. These tools and a self-contained implementation of the pipeline are freely available for non-commercial use at https://github.com/AllenInstitute/BarWare-pipeline .
基于条形码的多重方法可用于增加高通量和减少大型单细胞基因组学研究中的批次效应。尽管在样本采集和规模的灵活性方面具有优势,但在将每个细胞分配到其原始样本所需的数据解卷积步骤中存在额外的复杂性。
为了满足高效样本解卷积的计算需求,我们开发了 BarCounter 和 BarMixer 这两个工具,它们分别计算条形码计数和解卷积混合单细胞数据成特定于样本的文件。这两个工具共同构成了 BarWare 管道,以支持从具有许多哈希 10x Genomics scRNA-seq 数据孔的大型测序项目中进行多路复用。
BarWare 是一组通过 shell 脚本链接的模块化工具:BarCounter 是一种用 C 语言实现的计算高效的条形码序列定量工具;BarMixer 是一个用于识别条形码群体、合并来自多个孔的条形码数据以及与 scRNA-seq 数据相关的质量控制报告的 R 包。这些工具和管道的自包含实现可在非商业用途上免费获得,网址为 https://github.com/AllenInstitute/BarWare-pipeline。