Department of Computer Science, Johns Hopkins University, Baltimore, MD, USA.
Department of Biology, Johns Hopkins University, Baltimore, MD, USA.
Science. 2022 Apr;376(6588):eabl3533. doi: 10.1126/science.abl3533. Epub 2022 Apr 1.
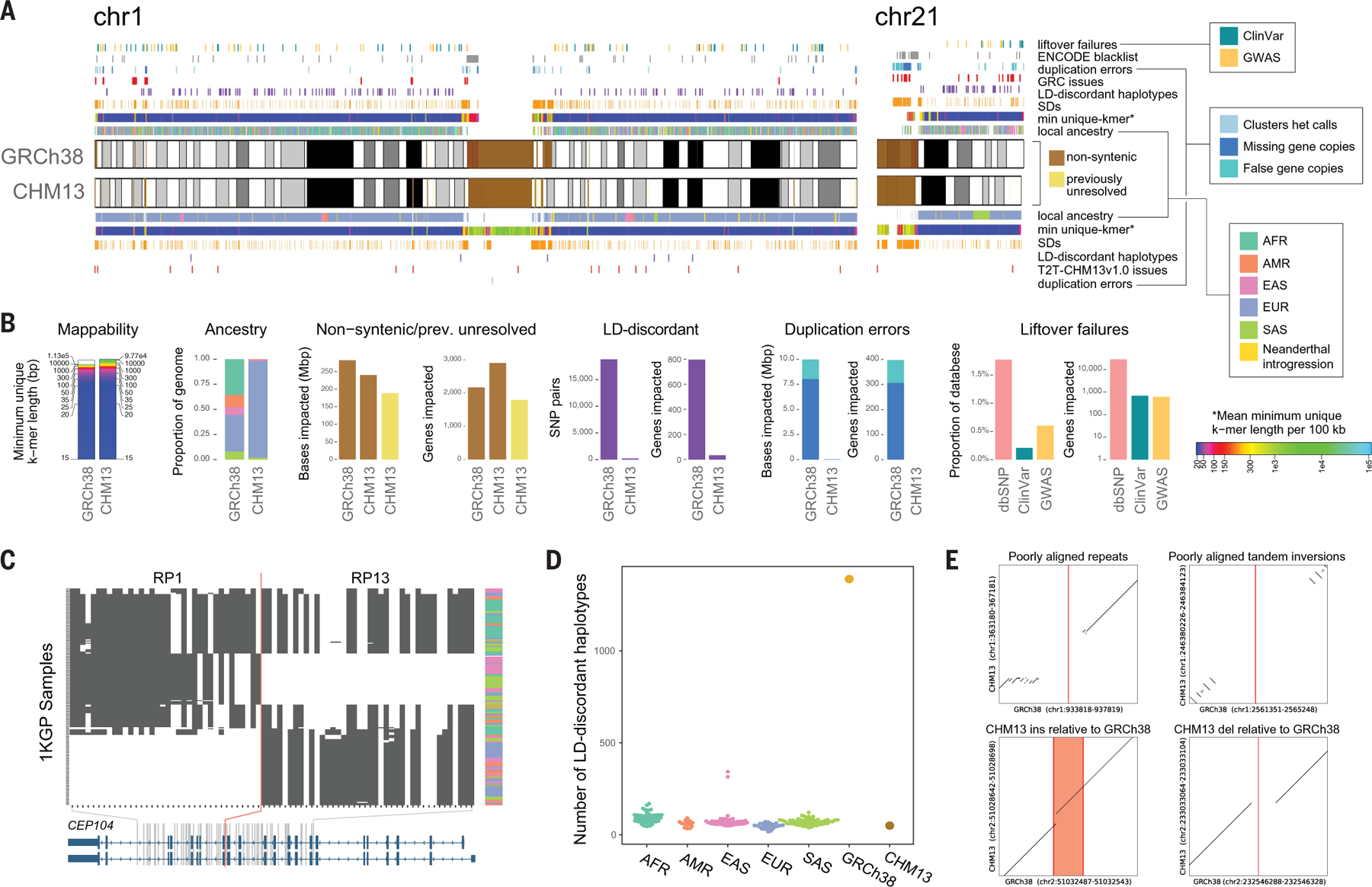
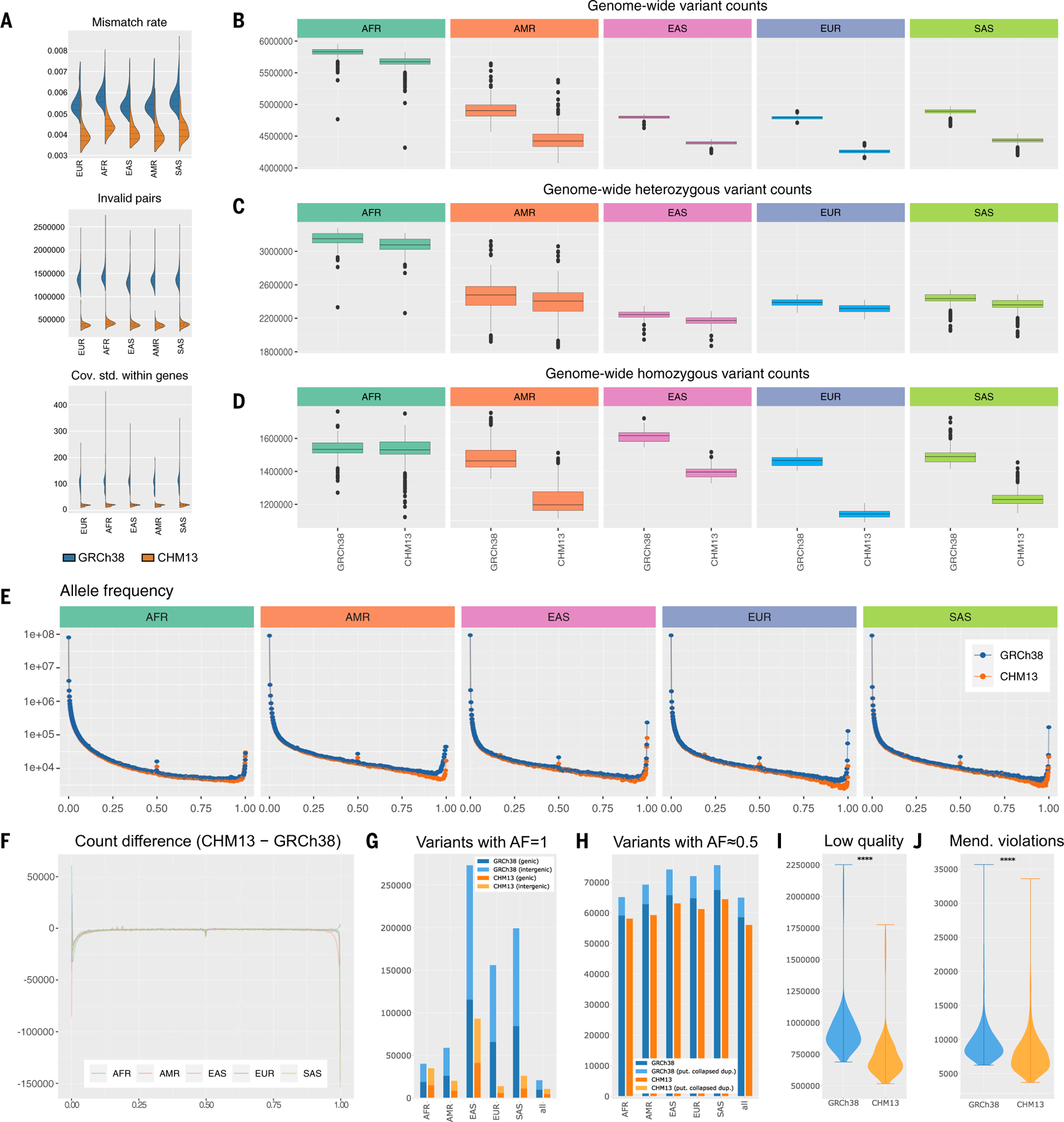
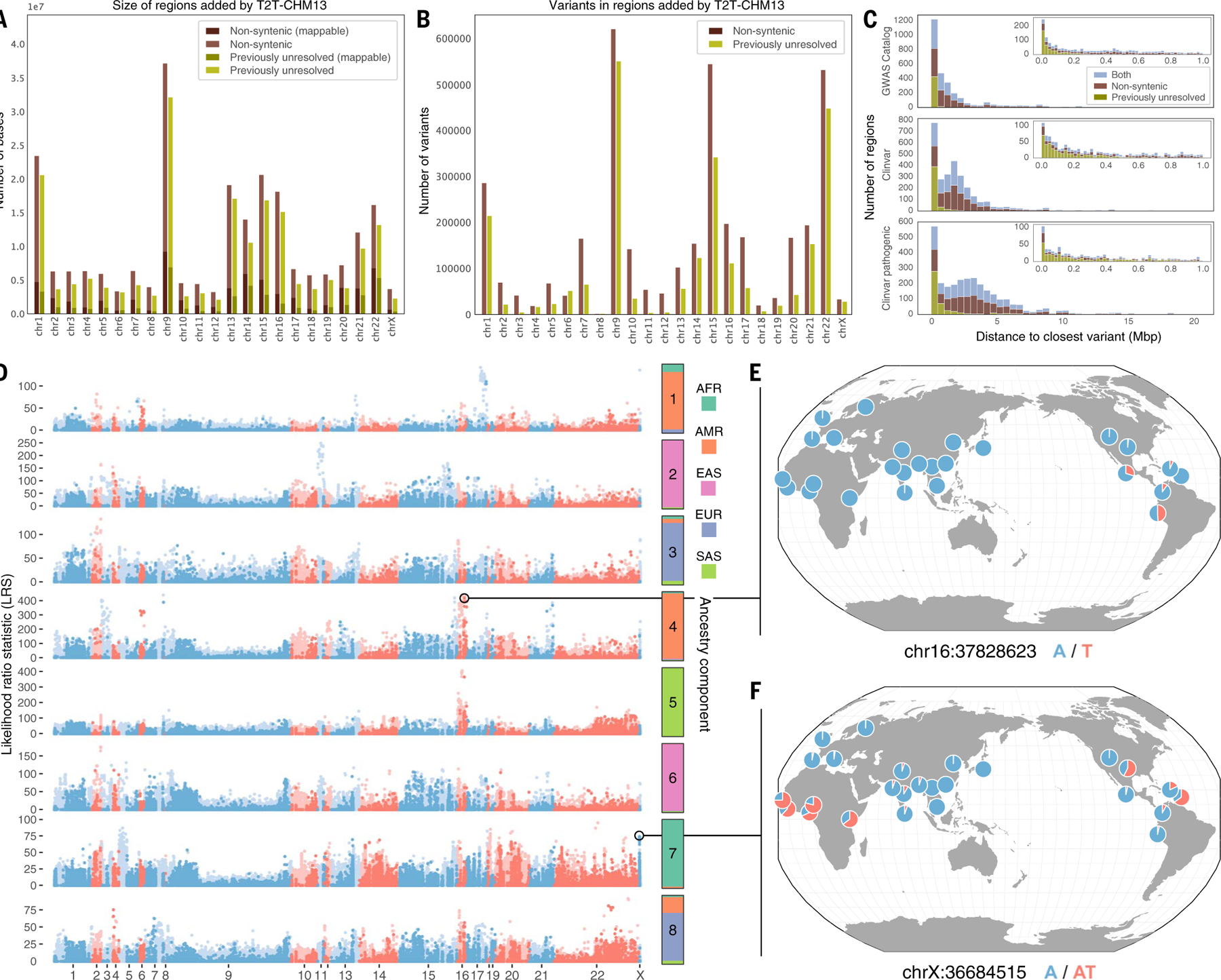
Compared to its predecessors, the Telomere-to-Telomere CHM13 genome adds nearly 200 million base pairs of sequence, corrects thousands of structural errors, and unlocks the most complex regions of the human genome for clinical and functional study. We show how this reference universally improves read mapping and variant calling for 3202 and 17 globally diverse samples sequenced with short and long reads, respectively. We identify hundreds of thousands of variants per sample in previously unresolved regions, showcasing the promise of the T2T-CHM13 reference for evolutionary and biomedical discovery. Simultaneously, this reference eliminates tens of thousands of spurious variants per sample, including reduction of false positives in 269 medically relevant genes by up to a factor of 12. Because of these improvements in variant discovery coupled with population and functional genomic resources, T2T-CHM13 is positioned to replace GRCh38 as the prevailing reference for human genetics.
与之前的版本相比,端粒到端粒 CHM13 基因组增加了近 2 亿个碱基对的序列,纠正了数千个结构错误,并为人类基因组的最复杂区域解锁,以进行临床和功能研究。我们展示了这种参考如何普遍提高了对分别使用短读长和长读长测序的 3202 个和 17 个全球多样化样本的读取映射和变异调用。我们在以前无法解决的区域中为每个样本识别出数十万种变体,展示了 T2T-CHM13 参考在进化和生物医学发现方面的潜力。同时,该参考消除了每个样本中数万种虚假变体,包括在 269 个与医学相关的基因中,将假阳性减少了高达 12 倍。由于在变体发现方面的这些改进,以及人群和功能基因组资源,T2T-CHM13 有望取代 GRCh38 成为人类遗传学的主要参考。