Shi Wentao, Singha Manali, Srivastava Gopal, Pu Limeng, Ramanujam J, Brylinski Michal
Division of Electrical and Computer Engineering, Louisiana State University, Baton Rouge, LA, United States.
Department of Biological Sciences, Louisiana State University, Baton Rouge, LA, United States.
Front Pharmacol. 2022 Mar 11;13:837715. doi: 10.3389/fphar.2022.837715. eCollection 2022.
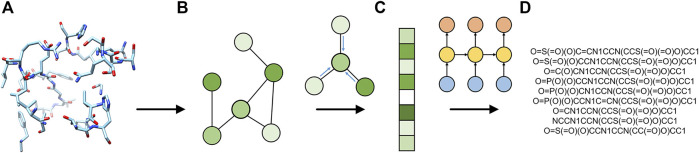
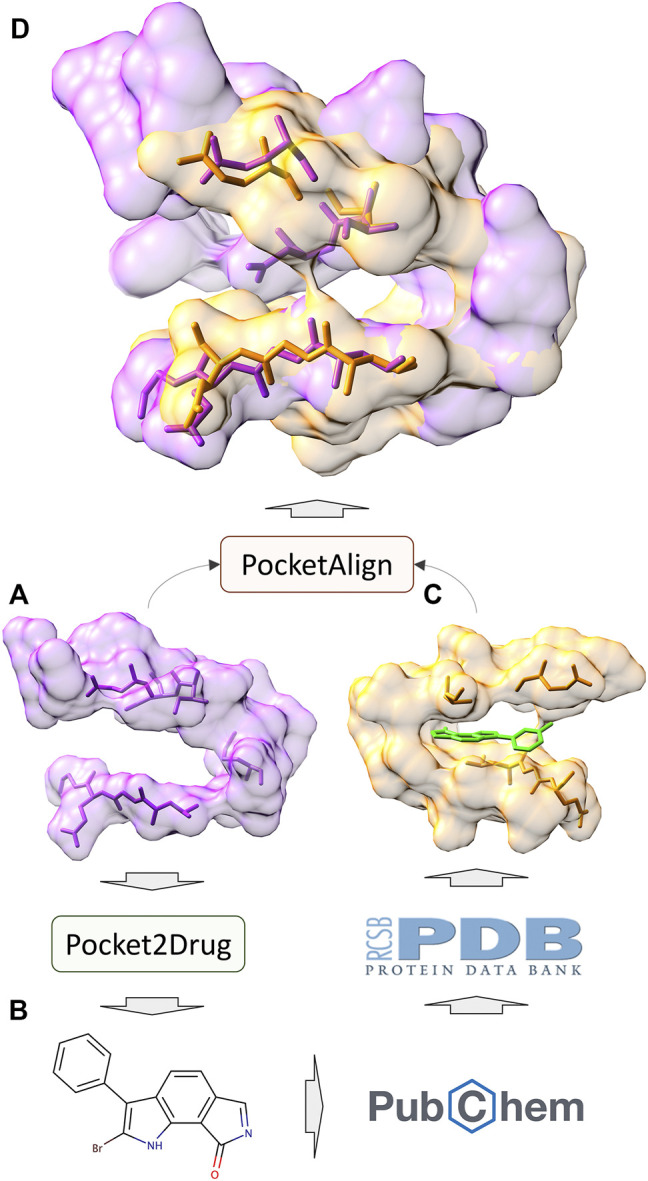
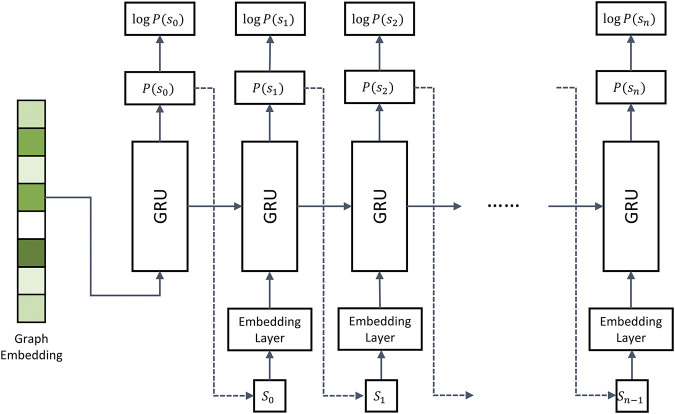
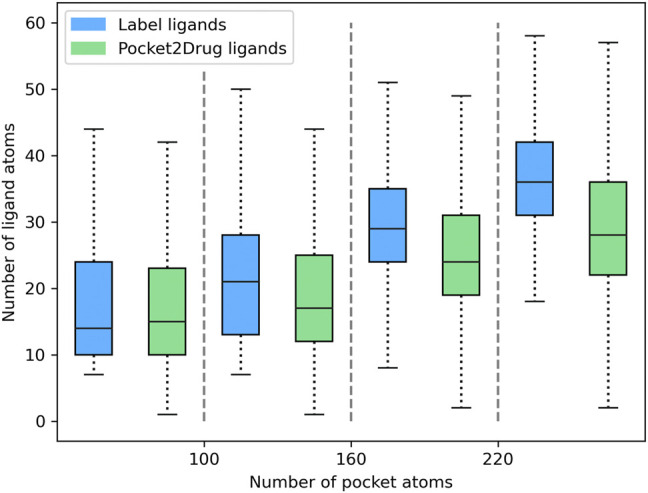
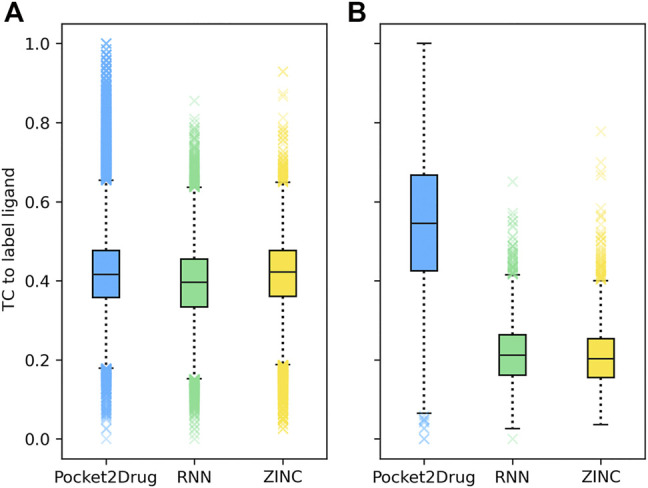
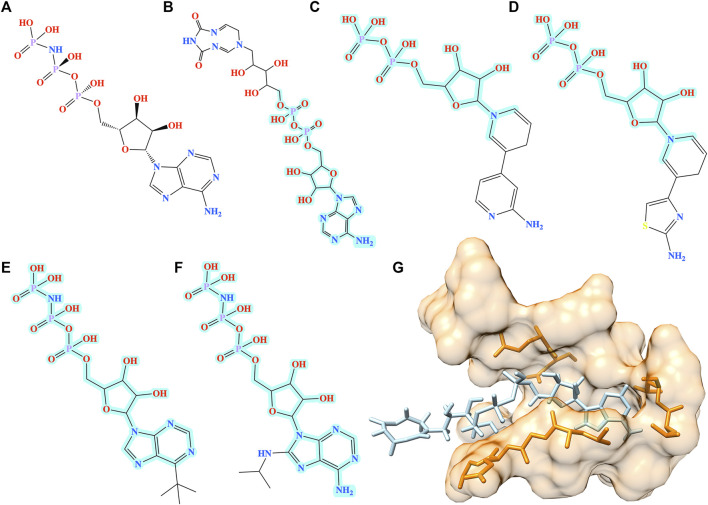
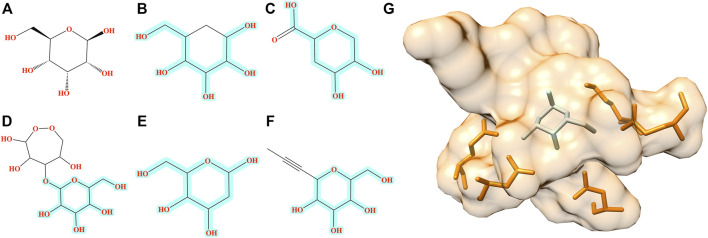
Computational modeling is an essential component of modern drug discovery. One of its most important applications is to select promising drug candidates for pharmacologically relevant target proteins. Because of continuing advances in structural biology, putative binding sites for small organic molecules are being discovered in numerous proteins linked to various diseases. These valuable data offer new opportunities to build efficient computational models predicting binding molecules for target sites through the application of data mining and machine learning. In particular, deep neural networks are powerful techniques capable of learning from complex data in order to make informed drug binding predictions. In this communication, we describe Pocket2Drug, a deep graph neural network model to predict binding molecules for a given a ligand binding site. This approach first learns the conditional probability distribution of small molecules from a large dataset of pocket structures with supervised training, followed by the sampling of drug candidates from the trained model. Comprehensive benchmarking simulations show that using Pocket2Drug significantly improves the chances of finding molecules binding to target pockets compared to traditional drug selection procedures. Specifically, known binders are generated for as many as 80.5% of targets present in the testing set consisting of dissimilar data from that used to train the deep graph neural network model. Overall, Pocket2Drug is a promising computational approach to inform the discovery of novel biopharmaceuticals.
计算建模是现代药物发现的重要组成部分。其最重要的应用之一是为药理学相关的靶蛋白选择有前景的候选药物。由于结构生物学的不断进步,在与各种疾病相关的众多蛋白质中发现了小分子的假定结合位点。这些宝贵的数据为通过应用数据挖掘和机器学习建立预测靶位点结合分子的高效计算模型提供了新机会。特别是,深度神经网络是强大的技术,能够从复杂数据中学习,以便做出明智的药物结合预测。在本通讯中,我们描述了Pocket2Drug,一种深度图神经网络模型,用于预测给定配体结合位点的结合分子。该方法首先通过监督训练从口袋结构的大数据集中学习小分子的条件概率分布,然后从训练模型中对候选药物进行采样。全面的基准模拟表明,与传统药物选择程序相比,使用Pocket2Drug显著提高了找到与靶口袋结合的分子的机会。具体而言,在由与用于训练深度图神经网络模型的数据不同的数据组成的测试集中,多达80.5%的靶标生成了已知的结合剂。总体而言,Pocket2Drug是一种有前景的计算方法,可为新型生物制药的发现提供信息。