Division of Electrical & Computer Engineering, Louisiana State University, Baton Rouge, LA, United States of America.
Department of Biological Sciences, Louisiana State University, Baton Rouge, LA, United States of America.
PLoS Comput Biol. 2019 Feb 4;15(2):e1006718. doi: 10.1371/journal.pcbi.1006718. eCollection 2019 Feb.
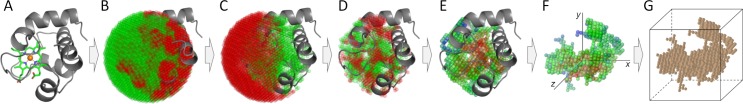
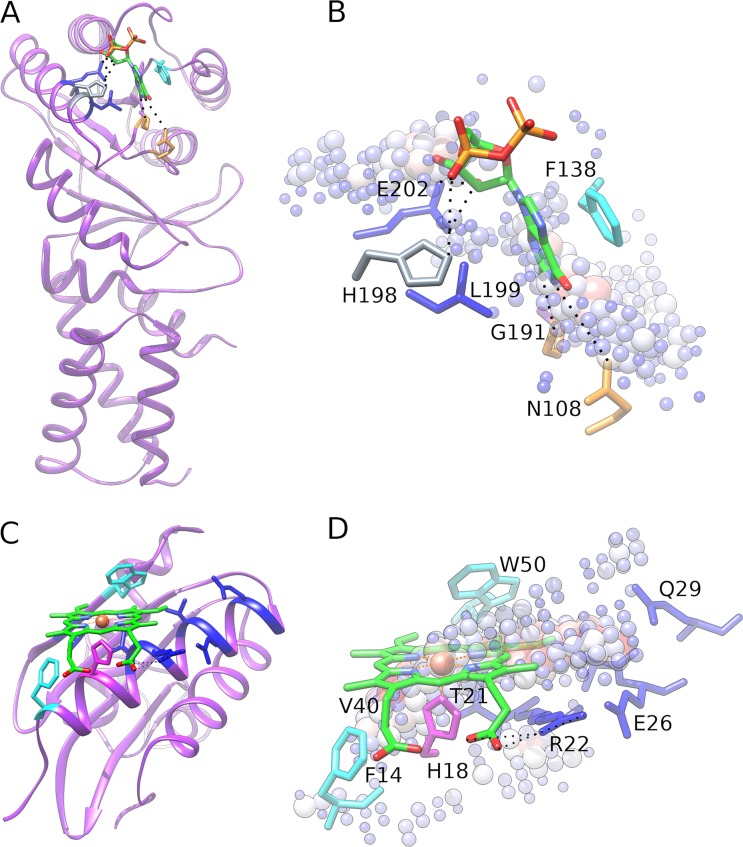
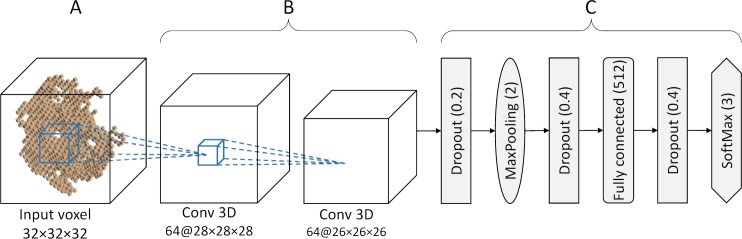
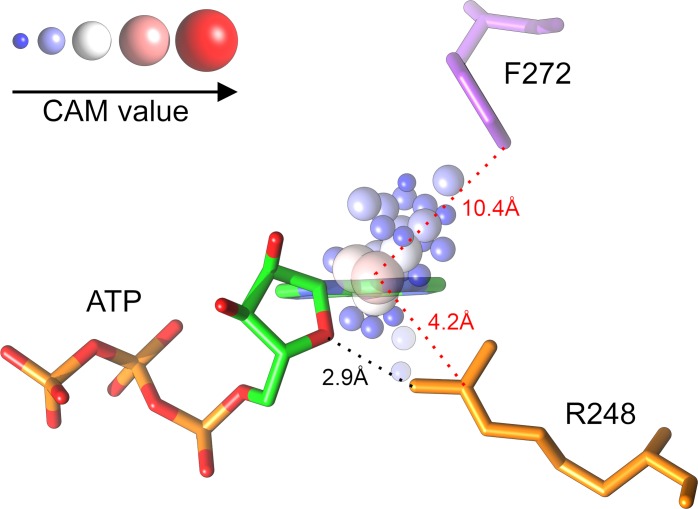
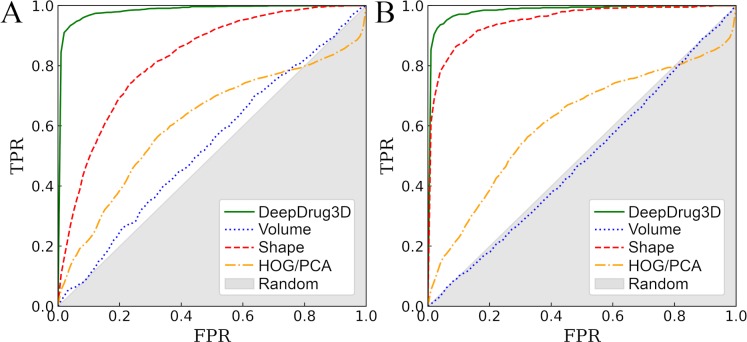
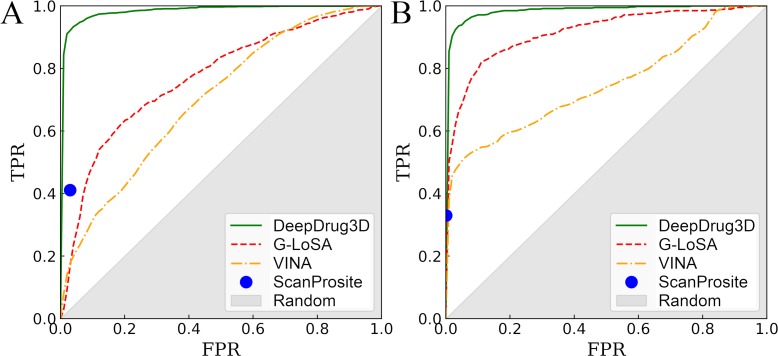
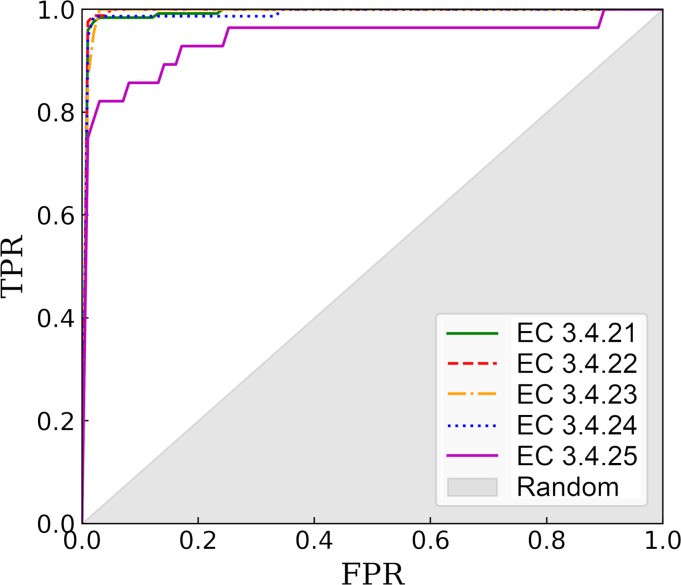
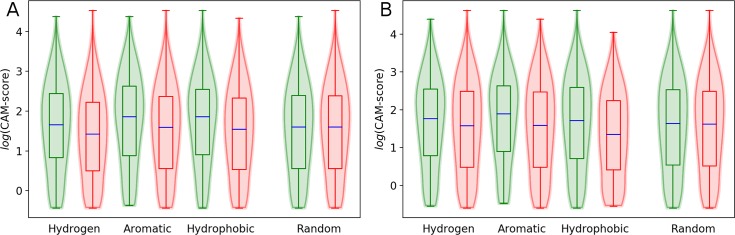
Comprehensive characterization of ligand-binding sites is invaluable to infer molecular functions of hypothetical proteins, trace evolutionary relationships between proteins, engineer enzymes to achieve a desired substrate specificity, and develop drugs with improved selectivity profiles. These research efforts pose significant challenges owing to the fact that similar pockets are commonly observed across different folds, leading to the high degree of promiscuity of ligand-protein interactions at the system-level. On that account, novel algorithms to accurately classify binding sites are needed. Deep learning is attracting a significant attention due to its successful applications in a wide range of disciplines. In this communication, we present DeepDrug3D, a new approach to characterize and classify binding pockets in proteins with deep learning. It employs a state-of-the-art convolutional neural network in which biomolecular structures are represented as voxels assigned interaction energy-based attributes. The current implementation of DeepDrug3D, trained to detect and classify nucleotide- and heme-binding sites, not only achieves a high accuracy of 95%, but also has the ability to generalize to unseen data as demonstrated for steroid-binding proteins and peptidase enzymes. Interestingly, the analysis of strongly discriminative regions of binding pockets reveals that this high classification accuracy arises from learning the patterns of specific molecular interactions, such as hydrogen bonds, aromatic and hydrophobic contacts. DeepDrug3D is available as an open-source program at https://github.com/pulimeng/DeepDrug3D with the accompanying TOUGH-C1 benchmarking dataset accessible from https://osf.io/enz69/.
配体结合位点的全面特征对于推断假设蛋白质的分子功能、追踪蛋白质之间的进化关系、设计具有所需底物特异性的酶以及开发具有改善选择性特征的药物都是非常有价值的。这些研究工作面临着重大的挑战,因为在不同的折叠结构中经常会观察到相似的口袋,这导致在系统水平上配体-蛋白质相互作用具有高度的混杂性。因此,需要开发新的算法来准确地分类结合位点。深度学习由于在广泛的学科领域中的成功应用而引起了极大的关注。在本通讯中,我们提出了 DeepDrug3D,这是一种使用深度学习来描述和分类蛋白质中结合口袋的新方法。它采用了一种最先进的卷积神经网络,其中生物分子结构被表示为体素,这些体素被赋予基于相互作用能的属性。目前的 DeepDrug3D 实现,经过训练可以检测和分类核苷酸和血红素结合位点,不仅实现了 95%的高精度,而且还具有推广到未见数据的能力,如类固醇结合蛋白和肽酶的验证。有趣的是,对结合口袋的强判别区域的分析表明,这种高精度的分类是通过学习特定分子相互作用的模式(如氢键、芳香族和疏水性接触)而产生的。DeepDrug3D 可作为开源程序在 https://github.com/pulimeng/DeepDrug3D 上获得,其附带的 TOUGH-C1 基准数据集可从 https://osf.io/enz69/ 获得。