Yun J Winny, Washington Caretia, McCormick Joi, Stevenson Emily, Alexander J Steven
Department of Molecular and Cellular Physiology, LSUHSC, Shreveport, LA 71103, USA.
Pathophysiology. 2021 Feb 11;28(1):64-75. doi: 10.3390/pathophysiology28010006.
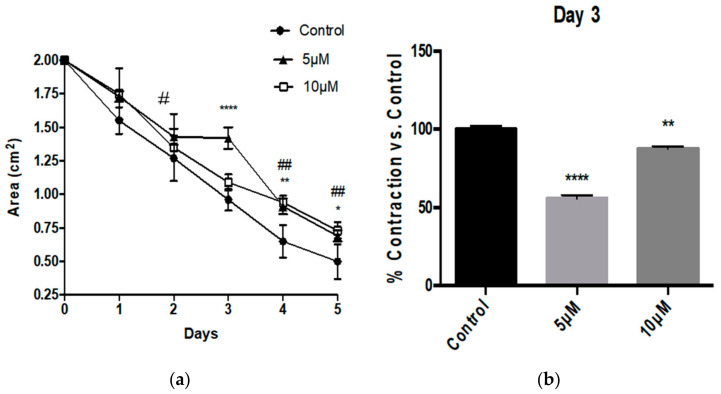
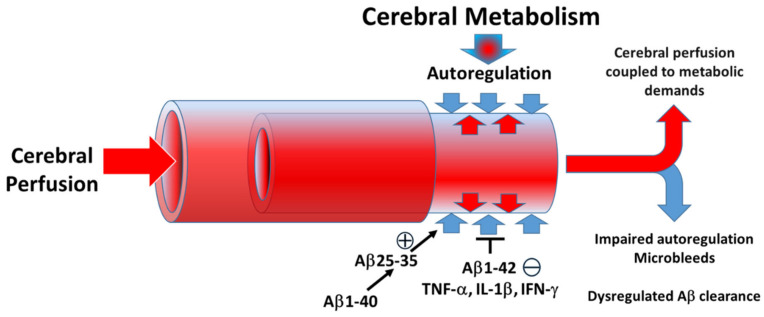
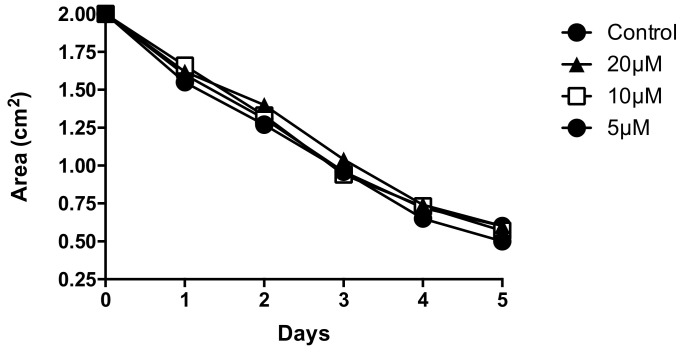
Alzheimer's Disease (AD) is a neurodegenerative condition characterized both by the presence of tau protein neurofibrillary tangles and amyloid beta (Aβ) containing extracellular "plaques". The cleavage of amyloid precursor protein (APP) yields several Aβ peptides. Although Aβ toxicity to neurons has been described extensively, its effects on other components of the neurovasculature such as vascular smooth muscle cells have been less well characterized. AD is now also recognized as a neurovascular disease characterized by cerebral microbleeds and disturbances in autoregulation. AD is also a neuroinflammatory condition in which several proinflammatory cytokines are elevated and may contribute to the intensification of AD severity. Cerebral autoregulation (the mechanism by which brain blood flow is maintained despite changes in perfusion pressure) is extremely tightly controlled in the brain and shows disturbances in AD. The failure of autoregulation in AD may make the brain susceptible to cerebral microbleeds through a reduced capacity to limit blood flow when pressure is increased. Conversely, reduced vasodilation during low flow might could also exacerbate tissue hypoxia. Currently, whether and how Aβ peptides and inflammatory cytokines depress brain smooth muscle cell tonic contraction is not known, but could reveal important targets in the preservation of autoregulation which is disturbed in AD. We used a collagen gel contractility assay to evaluate the influence of Aβ25-35, Aβ1-40 and Aβ1-42 peptides and inflammatory cytokines on the tonic contractility of human brain vascular smooth muscle cells (HBVSMC) as an in vitro model of cerebral autoregulation. We found that 5 and 10 μM Aβ1-42 significantly depressed HBVSM contractility, while Aβ1-40 5-20 μM had no effect on contractility. Conversely, Aβ25-35 (1-50 μM) increased contractility. Interestingly, the inflammatory cytokines TNF-α (20 ng/mL), IL-1β (20 ng/mL) and IFN-γ (1000 U/mL) also depressed HBVSM tonic contractility alone and in combination. These data suggest that both the inflammatory milieu in AD as well as the abundance of Aβ peptides may promote autoregulatory failure and increase brain susceptibility to dysregulated perfusion and microbleeds which are an important and devastating characteristic of AD.
阿尔茨海默病(AD)是一种神经退行性疾病,其特征在于存在tau蛋白神经原纤维缠结和含有细胞外“斑块”的β-淀粉样蛋白(Aβ)。淀粉样前体蛋白(APP)的切割产生几种Aβ肽。尽管Aβ对神经元的毒性已被广泛描述,但其对神经血管系统其他成分(如血管平滑肌细胞)的影响尚未得到充分表征。AD现在也被认为是一种神经血管疾病,其特征为脑微出血和自身调节紊乱。AD也是一种神经炎症性疾病,其中几种促炎细胞因子升高,可能导致AD严重程度加剧。脑自身调节(尽管灌注压力发生变化仍能维持脑血流量的机制)在大脑中受到极其严格的控制,并且在AD中表现出紊乱。AD中自身调节的失败可能使大脑在压力增加时通过限制血流量的能力降低而容易发生脑微出血。相反,在低流量期间血管舒张减少也可能加剧组织缺氧。目前,尚不清楚Aβ肽和炎性细胞因子是否以及如何抑制脑平滑肌细胞的紧张性收缩,但这可能揭示在AD中受到干扰的自身调节的保存中的重要靶点。我们使用胶原凝胶收缩性测定法来评估Aβ25-35、Aβ1-40和Aβ1-42肽以及炎性细胞因子对人脑血管平滑肌细胞(HBVSMC)紧张性收缩的影响,将其作为脑自身调节的体外模型。我们发现5和10μM的Aβ1-42显著降低HBVSM收缩性,而5-20μM的Aβ1-40对收缩性没有影响。相反,Aβ25-35(1-50μM)增加收缩性。有趣的是,炎性细胞因子TNF-α(20 ng/mL)、IL-1β(20 ng/mL)和IFN-γ(1000 U/mL)单独或联合使用也会降低HBVSM紧张性收缩。这些数据表明,AD中的炎性环境以及Aβ肽的丰度可能会促进自身调节失败,并增加大脑对灌注失调和微出血的易感性,而灌注失调和微出血是AD的一个重要且具有破坏性的特征。