Wu Zongkai, Wei Wanyi, Fan Hongzhen, Gu Yongsheng, Li Litao, Wang Hebo
Department of Neurology, Hebei Medical University, Shijiazhuang, China.
Department of Neurology, Hebei General Hospital, Shijiazhuang, China.
Front Genet. 2022 Mar 25;13:833545. doi: 10.3389/fgene.2022.833545. eCollection 2022.
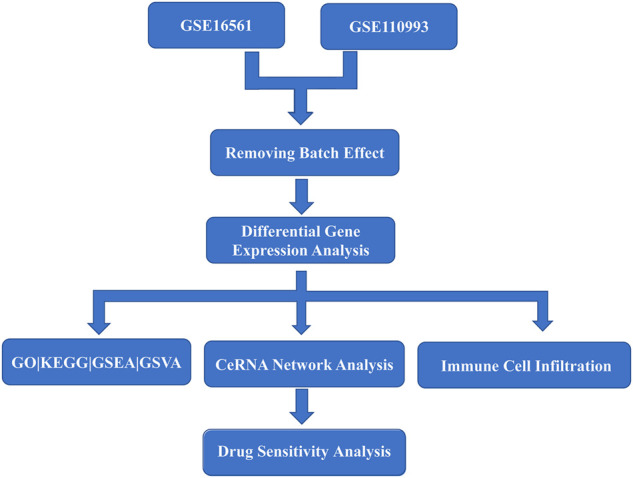
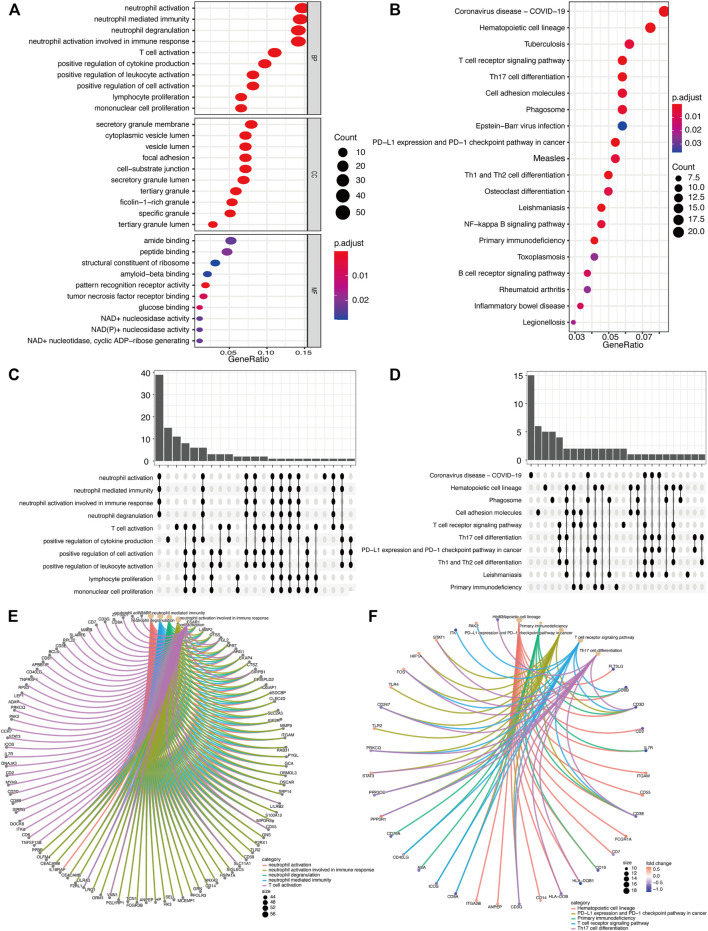
Acute ischemic stroke (AIS) is a severe neurological disease with complex pathophysiology, resulting in the disability and death. The goal of this study is to explore the underlying molecular mechanisms of AIS and search for new potential biomarkers and therapeutic targets. Integrative analysis of mRNA and miRNA profiles downloaded from Gene Expression Omnibus (GEO) was performed. We explored differentially expressed genes (DEGs) and differentially expressed miRNAs (DEMirs) after AIS. Target mRNAs of DEMirs and target miRNAs of DEGs were predicted with target prediction tools, and the intersections between DEGs and target genes were determined. Subsequently, Gene Ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) pathway enrichment analyses, Gene set enrichment analysis (GSEA), Gene set variation analysis (GSVA), competitive endogenous RNA (ceRNA) (lncRNA-miRNA-mRNA) network, protein-protein interaction (PPI) network, and gene transcription factors (TFs) network analyses were performed to identify hub genes and associated pathways. Furthermore, we obtained AIS samples with evaluation of immune cell infiltration and used CIBERSORT to determine the relationship between the expression of hub genes and infiltrating immune cells. Finally, we used the Genomics of Drug Sensitivity in Cancer (GDSC) database to predict the effect of the identified targets on drug sensitivity. We identified 293 DEGs and 26 DEMirs associated with AIS. DEGs were found to be mainly enriched in inflammation and immune-related signaling pathways through enrichment analysis. The ceRNA network included nine lncRNAs, 13 miRNAs, and 21 mRNAs. We used the criterion AUC >0.8, to screen a 3-gene signature (FBL, RPS3, and RPS15) and the aberrantly expressed miRNAs (hsa-miR-125a-5p, hsa-miR-125b-5p, hsa-miR-148b-3p, and hsa-miR-143-3p) in AIS, which were verified by a method of quantitative PCR (qPCR) in HT22 cells. T cells CD8, B cells naïve, and activated NK cells had statistical increased in number compared with the acute cerebral infarction group. By predicting the IC50 of the patient to the drug, AZD0530, Z.LLNle.CHO and NSC-87877 with significant differences between the groups were screened out. AIS demonstrated heterogeneity in immune infiltrates that correlated with the occurrence and development of diseases. These findings may contribute to a better understanding of the molecular mechanisms of AIS and provide the basis for the development of novel treatment targets in AIS.
急性缺血性卒中(AIS)是一种具有复杂病理生理学的严重神经系统疾病,可导致残疾和死亡。本研究的目的是探索AIS潜在的分子机制,并寻找新的潜在生物标志物和治疗靶点。我们对从基因表达综合数据库(GEO)下载的mRNA和miRNA谱进行了综合分析。我们探究了AIS后差异表达基因(DEGs)和差异表达miRNA(DEMirs)。使用靶标预测工具预测DEMirs的靶标mRNA和DEGs的靶标miRNA,并确定DEGs与靶标基因之间的交集。随后,进行基因本体(GO)和京都基因与基因组百科全书(KEGG)通路富集分析、基因集富集分析(GSEA)、基因集变异分析(GSVA)、竞争性内源性RNA(ceRNA)(lncRNA-miRNA-mRNA)网络、蛋白质-蛋白质相互作用(PPI)网络和基因转录因子(TFs)网络分析,以鉴定枢纽基因和相关通路。此外,我们获取了评估免疫细胞浸润的AIS样本,并使用CIBERSORT确定枢纽基因表达与浸润免疫细胞之间的关系。最后,我们使用癌症药物敏感性基因组学(GDSC)数据库预测已鉴定靶点对药物敏感性的影响。我们鉴定出293个与AIS相关的DEGs和26个DEMirs。通过富集分析发现DEGs主要富集于炎症和免疫相关信号通路。ceRNA网络包括9个lncRNA、13个miRNA和21个mRNA。我们使用AUC>0.8的标准,筛选出AIS中的一个3基因特征(FBL、RPS3和RPS15)以及异常表达的miRNA(hsa-miR-125a-5p、hsa-miR-125b-5p、hsa-miR-148b-3p和hsa-miR-143-3p),并在HT22细胞中通过定量PCR(qPCR)方法进行了验证。与急性脑梗死组相比,T细胞CD8、幼稚B细胞和活化NK细胞数量在统计学上显著增加。通过预测患者对药物AZD0530、Z.LLNle.CHO和NSC-87877的半数抑制浓度(IC50),筛选出组间有显著差异的药物。AIS在免疫浸润方面表现出异质性,这与疾病的发生和发展相关。这些发现可能有助于更好地理解AIS的分子机制,并为AIS新治疗靶点的开发提供依据。