Fang Yuan, Abuduxikuer Kuerbanjiang, Wang Yi-Zhen, Li Shao-Mei, Chen Lian, Wang Jian-She
Department of Pathology, Anhui Provincial Children's Hospital, Hefei, China.
Department of Hepatology, Children's Hospital of Fudan University, Shanghai, China.
Front Genet. 2022 Mar 24;13:833495. doi: 10.3389/fgene.2022.833495. eCollection 2022.
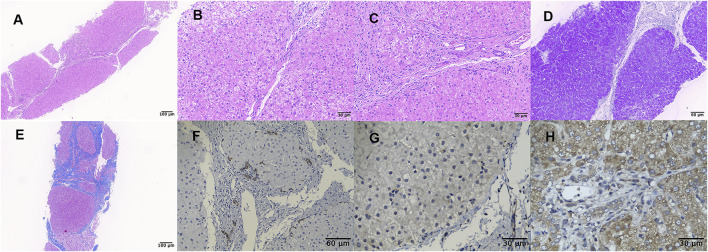
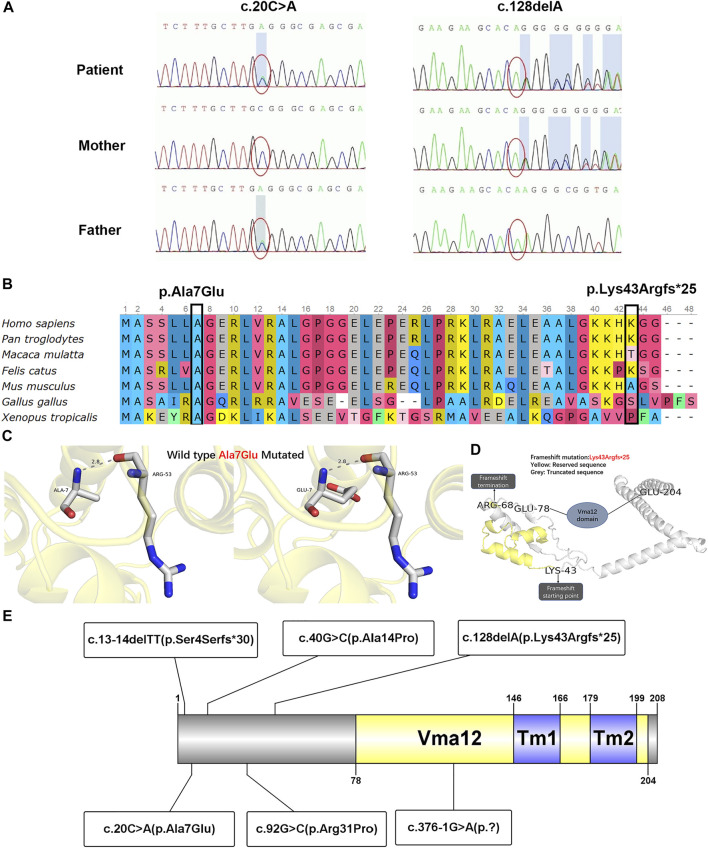
TMEM199-congenital disorder of glycosylation (TMEM199-CDG) is a rare autosomal recessive inherited disease characterized by chronically elevated serum transaminase, decreased serum ceruloplasmin, steatosis and/or fibrosis, mutation, reduced level of TMEM199 protein, and abnormal protein glycosylation. The information of a Chinese patient with TMEM199-CDG in the Children's Hospital of Fudan University was reviewed. The patient's clinical, pathological, and molecular features were obtained by clinical data study, liver biopsy, immunohistochemistry, and molecular genetic analysis. A 4-year-old Chinese boy presented with hypertransaminasemia, hypercholesterolemia, elevated alkaline phosphatase, decreased serum ceruloplasmin and serum copper level, and coagulopathy since birth. To the best of our knowledge, novel findings included strabismus, cirrhosis by liver biopsy, reduced expression of TMEM199 by immunohistochemistry, and a frameshift variant of c.128delA/p.Lys43Argfs*25 in the gene. This case added to the phenotypic and genotypic spectrum of TMEM199-CDG.
跨膜蛋白199相关先天性糖基化障碍(TMEM199-CDG)是一种罕见的常染色体隐性遗传病,其特征为血清转氨酶持续升高、血清铜蓝蛋白降低、脂肪变性和/或纤维化、基因突变、TMEM199蛋白水平降低以及蛋白糖基化异常。对复旦大学附属儿科医院一名TMEM199-CDG中国患者的信息进行了回顾。通过临床资料研究、肝活检、免疫组织化学和分子遗传学分析获得了该患者的临床、病理和分子特征。一名4岁中国男孩自出生以来就出现高转氨酶血症、高胆固醇血症、碱性磷酸酶升高、血清铜蓝蛋白和血清铜水平降低以及凝血功能障碍。据我们所知,新发现包括斜视、肝活检显示肝硬化、免疫组织化学检测显示TMEM199表达降低以及该基因存在c.128delA/p.Lys43Argfs*25移码变异。该病例丰富了TMEM199-CDG的表型和基因型谱。