Unit of Pediatrics, Department of Medicine, Surgery and Dentistry, Scuola Medica Salernitana, University of Salerno, Baronissi, (Sa), Italy.
Division of Pediatrics, Lund University, Lund, Sweden.
Orphanet J Rare Dis. 2018 Jan 10;13(1):4. doi: 10.1186/s13023-017-0757-3.
TMEM199 deficiency was recently shown in four patients to cause liver disease with steatosis, elevated serum transaminases, cholesterol and alkaline phosphatase and abnormal protein glycosylation. There is no information on the long-term outcome in this disorder.
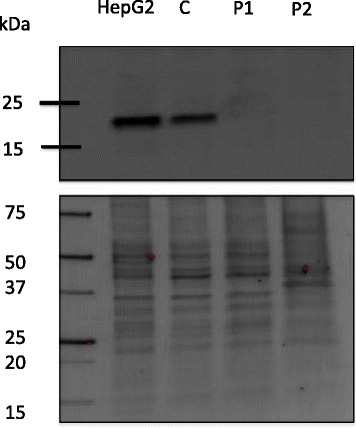
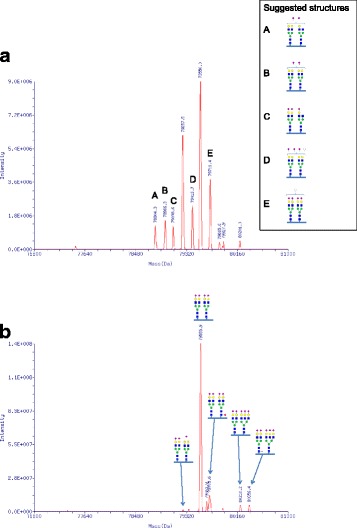
We here present three novel patients with TMEM199-CDG. All three patients carried the same set of mutations (c.13-14delTT (p.Ser4Serfs*30) and c.92G > C (p.Arg31Pro), despite only two were related (siblings). One mutation (c.92G > C) was described previously whereas the other was deemed pathogenic due to its early frameshift. Western Blot analysis confirmed a reduced level of TMEM199 protein in patient fibroblasts and all patients showed a similar glycosylation defect. The patients presented with a very similar clinical and biochemical phenotype to the initial publication, confirming that TMEM199-CDG is a non-encephalopathic liver disorder. Two of the patients were clinically assessed over two decades without deterioration.
A rising number of disorders affecting Golgi homeostasis have been published over the last few years. A hallmark finding is deficiency in protein glycosylation, both in N- and O-linked types. Most of these disorders have signs of both liver and brain involvement. However, the present and the four previously reported patients do not show encephalopathy but a chronic, non-progressive (over decades) liver disease with hypertransaminasemia and steatosis. This information is crucial for the patient/families and clinician at diagnosis, as it distinguishes it from other Golgi homeostasis disorders, in having a much more favorable course.
最近有四项研究表明,TMEM199 缺陷会导致脂肪变性、血清转氨酶、胆固醇和碱性磷酸酶升高以及蛋白质糖基化异常的肝脏疾病。目前尚无关于该疾病长期预后的信息。
我们在此介绍三位新的 TMEM199-CDG 患者。尽管只有两位患者是亲属关系(兄弟姐妹),但他们都携带相同的一组突变(c.13-14delTT [p.Ser4Serfs*30]和 c.92G>C [p.Arg31Pro])。其中一个突变(c.92G>C)之前有过描述,而另一个突变由于其早期移码被认为是致病性的。Western Blot 分析证实患者成纤维细胞中 TMEM199 蛋白水平降低,所有患者均表现出相似的糖基化缺陷。这些患者的临床表现和生化表型与最初的报道非常相似,证实了 TMEM199-CDG 是一种非脑病性肝脏疾病。其中两位患者在二十年的临床评估中没有病情恶化。
过去几年,越来越多的影响高尔基体稳态的疾病被报道。一个显著的发现是蛋白质糖基化缺陷,包括 N-和 O-连接类型。这些疾病大多数都有肝脏和大脑受累的迹象。然而,本研究和之前报道的四位患者没有表现出脑病,而是表现为慢性、非进行性(数十年)的肝脏疾病,伴有高转氨酶血症和脂肪变性。这些信息对于患者/家属和临床医生在诊断时非常重要,因为它与其他高尔基体稳态疾病区分开来,具有更有利的病程。