Centre of Medical Genetics, University of Antwerp and Antwerp University Hospital, Edegem, Belgium.
Department of Clinical Genetics, Radboud University Medical Center, Nijmegen, The Netherlands.
Hum Mutat. 2022 Jul;43(7):815-831. doi: 10.1002/humu.24383. Epub 2022 Apr 28.
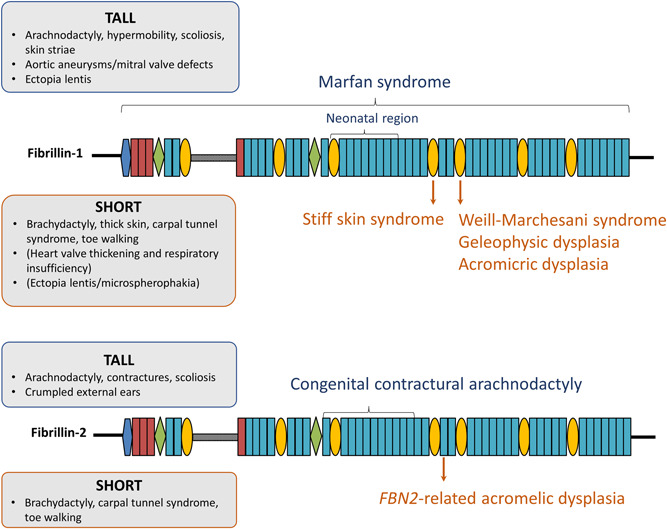
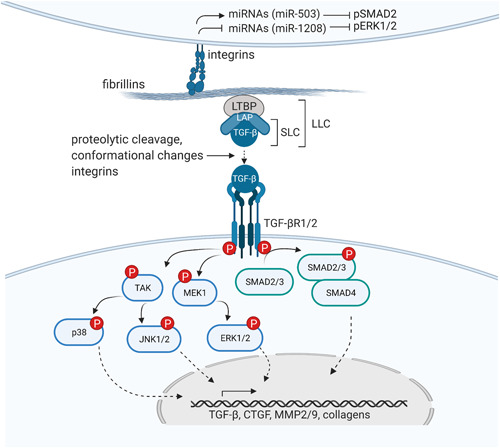
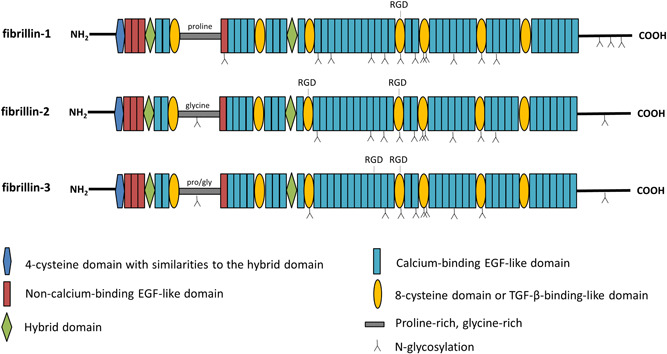
Different pathogenic variants in the fibrillin-1 gene (FBN1) cause Marfan syndrome and acromelic dysplasias. Whereas the musculoskeletal features of Marfan syndrome involve tall stature, arachnodactyly, joint hypermobility, and muscle hypoplasia, acromelic dysplasia patients present with short stature, brachydactyly, stiff joints, and hypermuscularity. Similarly, pathogenic variants in the fibrillin-2 gene (FBN2) cause either a Marfanoid congenital contractural arachnodactyly or a FBN2-related acromelic dysplasia that most prominently presents with brachydactyly. The phenotypic and molecular resemblances between both the FBN1 and FBN2-related disorders suggest that reciprocal pathomechanistic lessons can be learned. In this review, we provide an updated overview and comparison of the phenotypic and mutational spectra of both the "tall" and "short" fibrillinopathies. The future parallel functional study of both FBN1/2-related disorders will reveal new insights into how pathogenic fibrillin variants differently affect the fibrillin microfibril network and/or growth factor homeostasis in clinically opposite syndromes. This knowledge may eventually be translated into new therapeutic approaches by targeting or modulating the fibrillin microfibril network and/or the signaling pathways under its control.
不同的原纤维蛋白 1 基因(FBN1)致病变体导致马凡综合征和肢端骨发育不全。马凡综合征的骨骼肌肉特征包括身材高大、蜘蛛指(趾)、关节过度活动和肌肉萎缩,肢端骨发育不全患者则表现为身材矮小、短指(趾)、关节僵硬和肌肉过度发达。同样,原纤维蛋白 2 基因(FBN2)的致病变体导致马凡样先天性挛缩性蜘蛛指(趾)或 FBN2 相关肢端骨发育不全,最突出的表现为短指(趾)。FBN1 和 FBN2 相关疾病之间的表型和分子相似性表明,可以相互借鉴发病机制的经验。在这篇综述中,我们提供了 FBN1 和 FBN2 相关疾病的表型和突变谱的最新概述和比较。对这两种“高”和“矮”原纤维蛋白病的平行功能研究将揭示新的见解,了解致病原纤维蛋白变体如何以不同的方式影响临床上相反的综合征中的原纤维蛋白微纤维网络和/或生长因子动态平衡。这些知识最终可能通过靶向或调节原纤维蛋白微纤维网络及其控制的信号通路转化为新的治疗方法。