Zanganeh Pardis F, Barton Samantha K, Lim Katherine, Qian Elizabeth L, Crombie Duncan E, Bye Christopher R, Turner Bradley J
The Florey Institute of Neuroscience and Mental Health, University of Melbourne, Melbourne, VIC 3052, Australia.
The Perron Institute for Neurological and Translational Science, Queen Elizabeth Medical Centre, Nedlands, WA 6150, Australia.
Brain Commun. 2022 Mar 31;4(2):fcac081. doi: 10.1093/braincomms/fcac081. eCollection 2022.
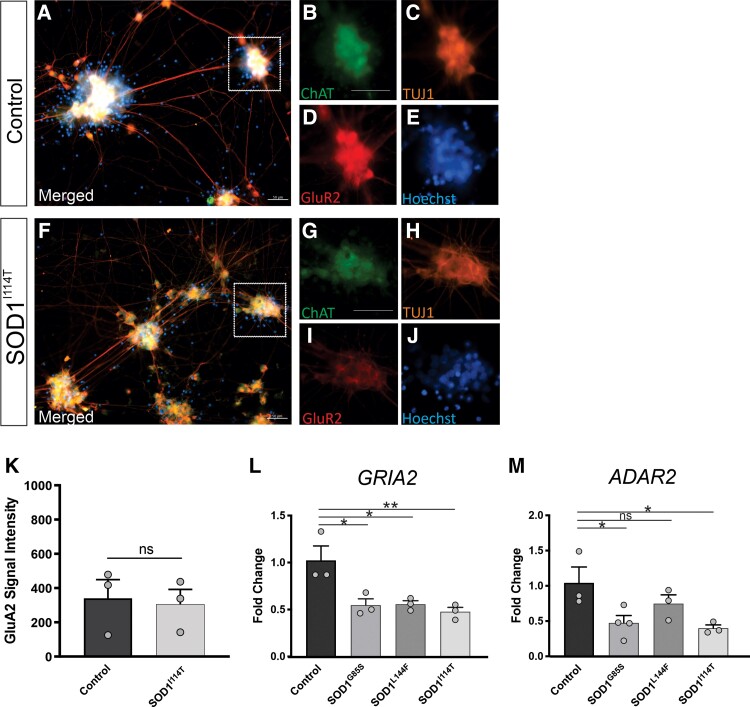
Amyotrophic lateral sclerosis is a late-onset adult neurodegenerative disease, although there is mounting electrophysiological and pathological evidence from patients and animal models for a protracted preclinical period of motor neuron susceptibility and dysfunction, long before clinical diagnosis. The key molecular mechanisms linked to motor neuron vulnerability in amyotrophic lateral sclerosis have been extensively studied using transcriptional profiling in motor neurons isolated from adult mutant superoxide dismutase 1 mice. However, neonatal and embryonic motor neurons from mutant superoxide dismutase 1 mice show abnormal morphology and hyperexcitability, suggesting preceding transcriptional dysregulation. Here, we used RNA sequencing on motor neurons isolated from embryonic superoxide dismutase 1 mice to determine the earliest molecular mechanisms conferring neuronal susceptibility and dysfunction known in a mouse model of amyotrophic lateral sclerosis. Transgenic superoxide dismutase 1 mice expressing the spinal motor neuron homeobox HB9:green fluorescent protein reporter allowed unambiguous identification and isolation of motor neurons using fluorescence-activated cell sorting. Gene expression profiling of isolated motor neurons revealed transcriptional dysregulation in superoxide dismutase 1 mice as early as embryonic Day 12.5 with the majority of differentially expressed genes involved in RNA processing and α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate-mediated glutamate receptor signalling. We confirmed dysregulation of the α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptor Subunit 2, at transcript and protein levels, in embryonic superoxide dismutase 1 mouse motor neurons and human motor neurons derived from amyotrophic lateral sclerosis patient induced pluripotent stem cells harbouring pathogenic superoxide dismutase 1 mutations. Mutant superoxide dismutase 1 induced pluripotent stem cell-derived motor neurons showed greater vulnerability to α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate-mediated excitotoxicity, consistent with α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptor Subunit 2 downregulation. Thus, α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptor Subunit 2 deficiency leading to enhanced α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptor signalling, calcium influx, hyperexcitability, and chronic excitotoxicity is a very early and intrinsic property of spinal motor neurons that may trigger amyotrophic lateral sclerosis pathogenesis later in life. This study reinforces the concept of therapeutic targeting of hyperexcitability and excitotoxicity as potential disease-modifying approaches for amyotrophic lateral sclerosis.
肌萎缩侧索硬化症是一种成年期发病较晚的神经退行性疾病,尽管来自患者和动物模型的电生理及病理学证据越来越多,表明在临床诊断之前很久,运动神经元就存在易感性和功能障碍的漫长临床前期。利用从成年突变型超氧化物歧化酶1小鼠分离出的运动神经元进行转录谱分析,已对与肌萎缩侧索硬化症中运动神经元易损性相关的关键分子机制进行了广泛研究。然而,突变型超氧化物歧化酶1小鼠的新生和胚胎运动神经元表现出形态异常和过度兴奋,提示存在先前的转录失调。在此,我们对从胚胎超氧化物歧化酶1小鼠分离出的运动神经元进行RNA测序,以确定在肌萎缩侧索硬化症小鼠模型中已知的赋予神经元易感性和功能障碍的最早分子机制。表达脊髓运动神经元同源框HB9:绿色荧光蛋白报告基因的转基因超氧化物歧化酶1小鼠,允许使用荧光激活细胞分选明确鉴定和分离运动神经元。对分离出的运动神经元进行基因表达谱分析发现,早在胚胎第12.5天,超氧化物歧化酶1小鼠就存在转录失调,大多数差异表达基因参与RNA加工和α-氨基-3-羟基-5-甲基-4-异恶唑丙酸介导的谷氨酸受体信号传导。我们在胚胎超氧化物歧化酶1小鼠运动神经元以及源自携带致病性超氧化物歧化酶1突变的肌萎缩侧索硬化症患者诱导多能干细胞的人运动神经元中,在转录和蛋白质水平证实了α-氨基-3-羟基-5-甲基-4-异恶唑丙酸受体亚基2的失调。突变型超氧化物歧化酶1诱导多能干细胞衍生的运动神经元对α-氨基-3-羟基-5-甲基-4-异恶唑丙酸介导的兴奋毒性表现出更大的易感性,这与α-氨基-3-羟基-5-甲基-4-异恶唑丙酸受体亚基2下调一致。因此,α-氨基-3-羟基-5-甲基-4-异恶唑丙酸受体亚基2缺乏导致α-氨基-3-羟基-5-甲基-4-异恶唑丙酸受体信号增强、钙内流、过度兴奋和慢性兴奋毒性,是脊髓运动神经元的一种非常早期的内在特性,可能在生命后期引发肌萎缩侧索硬化症的发病机制。这项研究强化了将过度兴奋和兴奋毒性作为肌萎缩侧索硬化症潜在疾病修饰方法进行治疗靶向的概念。