Han Maozhen, Zhang Na, Mao Yujie, Huang Bingbing, Ren Mengfei, Peng Zhangjie, Bai Zipeng, Chen Long, Liu Yan, Wang Shanshan, Huang Shenghai, Cheng Zhixiang
School of Life Sciences, Anhui Medical University, Hefei, China.
Department of Blood Transfusion, The Fourth Affiliated Hospital of Anhui Medical University, Hefei, China.
Front Microbiol. 2022 Apr 8;13:839015. doi: 10.3389/fmicb.2022.839015. eCollection 2022.
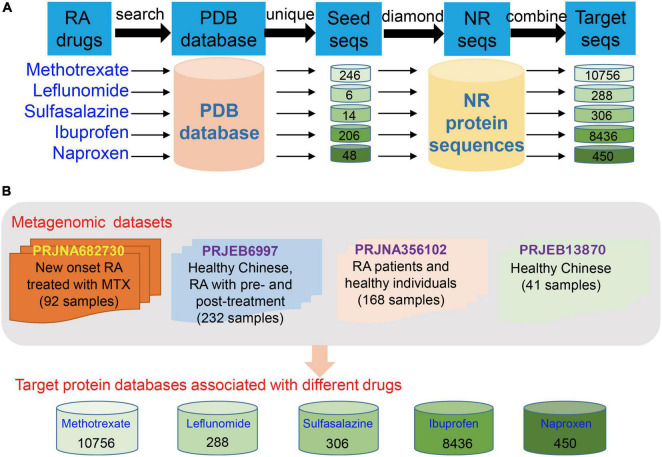
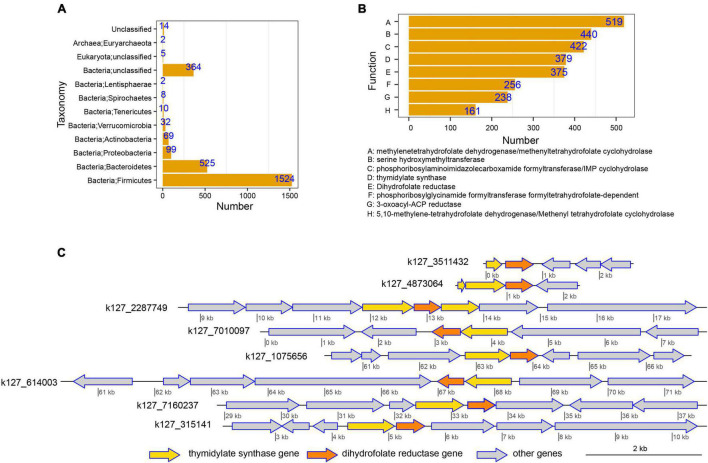
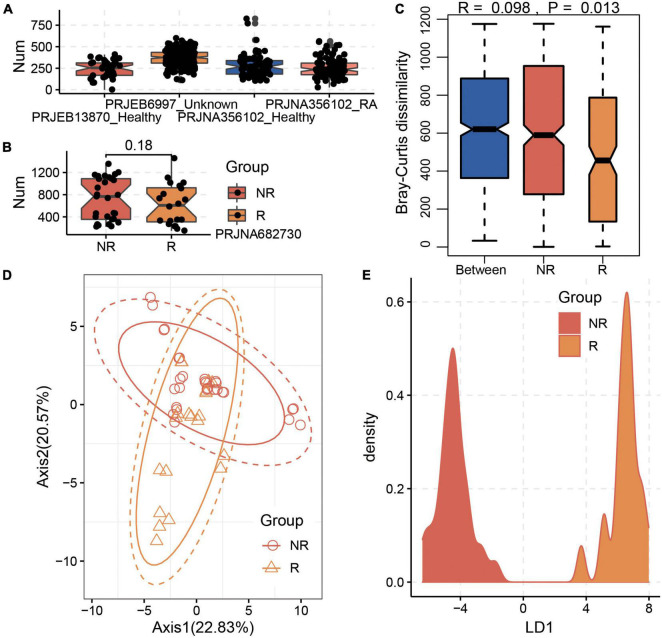
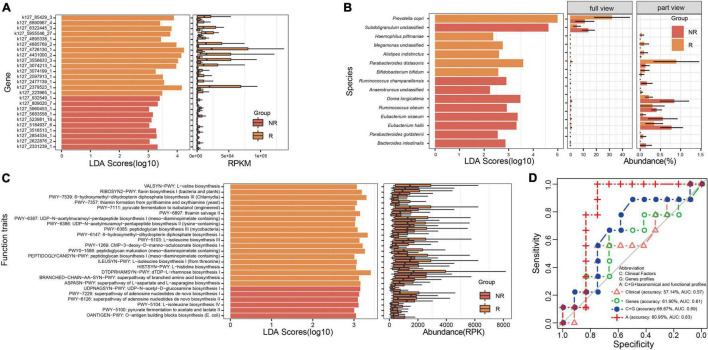
Gut microbiota plays an essential role in the development of rheumatoid arthritis (RA) and affects drug responses. However, the underlying mechanism remains elusive and urgent to elucidate to explore the pathology and clinical treatment of RA. Therefore, we selected methotrexate (MTX) as an example of RA drugs to explore the interactions between the gut microbiota and drug responses and obtain an in-depth understanding of their correlation from the perspective of the metabolic capability of gut microbiota on drug metabolism. We identified 2,654 proteins and the corresponding genes involved in MTX metabolism and then profiled their abundances in the gut microbiome datasets of four cohorts. We found that the gut microbiota harbored various genes involved in MTX metabolism in healthy individuals and RA patients. Interestingly, the number of genes involved in MTX metabolism was not significantly different between response (R) and non-response (NR) groups to MTX, but the gene composition in the microbial communities significantly differed between these two groups. Particularly, several models were built based on clinical information, as well as data on the gene, taxonomical, and functional biomarkers by using the random forest algorithm and then validated. Our findings provide bases for clinical management not only of RA but also other gut microbiome-related diseases. First, it suggests that the potential metabolic capability of gut microbiota on drug metabolism is important because they affect drug efficiency; as such, clinical treatment strategies should incorporate the gene compositions of gut microbial communities, in particular genes involved in drug metabolism. Second, a suitable model can be developed to determine hosts' responses to drugs before clinical treatment.
肠道微生物群在类风湿性关节炎(RA)的发展中起着至关重要的作用,并影响药物反应。然而,其潜在机制仍不清楚,迫切需要阐明以探索RA的病理学和临床治疗方法。因此,我们选择甲氨蝶呤(MTX)作为RA药物的例子,以探索肠道微生物群与药物反应之间的相互作用,并从肠道微生物群对药物代谢的代谢能力角度深入了解它们之间的相关性。我们鉴定了2654种参与MTX代谢的蛋白质及其相应基因,然后分析了它们在四个队列的肠道微生物组数据集中的丰度。我们发现,健康个体和RA患者的肠道微生物群中都含有多种参与MTX代谢的基因。有趣的是,对MTX有反应(R)和无反应(NR)的两组之间,参与MTX代谢的基因数量没有显著差异,但这两组微生物群落中的基因组成存在显著差异。特别是,利用随机森林算法基于临床信息以及基因、分类学和功能生物标志物的数据建立了几个模型,然后进行了验证。我们的研究结果为RA以及其他肠道微生物群相关疾病的临床管理提供了依据。首先,这表明肠道微生物群对药物代谢的潜在代谢能力很重要,因为它们会影响药物疗效;因此,临床治疗策略应纳入肠道微生物群落的基因组成,特别是参与药物代谢的基因。其次,可以开发一个合适的模型,在临床治疗前确定宿主对药物的反应。