Wang Meng, Cotter Edmund, Wang Ya-Juan, Fu Xu, Whittsette Angela L, Lynch Joseph W, Wiseman R Luke, Kelly Jeffery W, Keramidas Angelo, Mu Ting-Wei
Department of Physiology and Biophysics, Case Western Reserve University School of Medicine, 10900 Euclid Ave, Cleveland, OH, 44106, USA.
Queensland Brain Institute, the University of Queensland, Brisbane, QLD, 4072, Australia.
Cell Biosci. 2022 Apr 27;12(1):48. doi: 10.1186/s13578-022-00783-w.
Genetic variants in the subunits of the gamma-aminobutyric acid type A (GABA) receptors are implicated in the onset of multiple pathologic conditions including genetic epilepsy. Previous work showed that pathogenic GABA subunits promote misfolding and inefficient assembly of the GABA receptors, limiting receptor expression and activity at the plasma membrane. However, GABA receptors containing variant subunits can retain activity, indicating that enhancing the folding, assembly, and trafficking of these variant receptors offers a potential opportunity to mitigate pathology associated with genetic epilepsy.
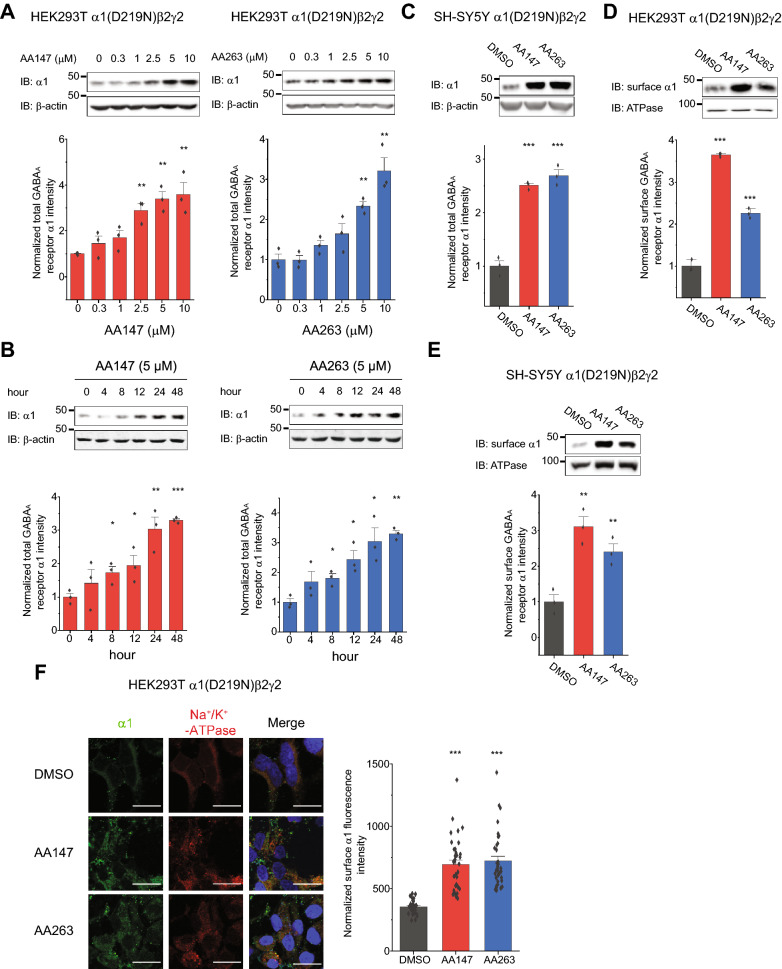
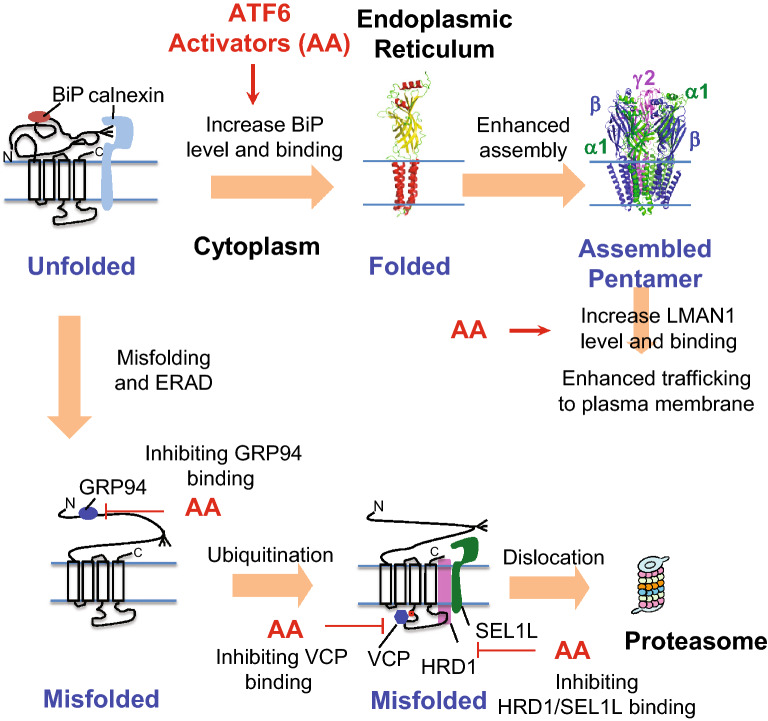
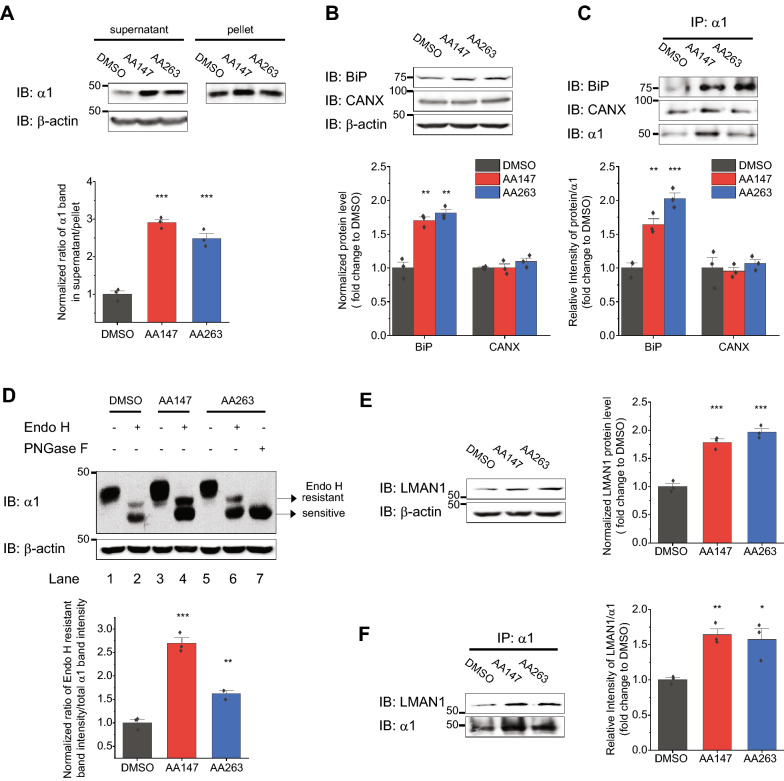
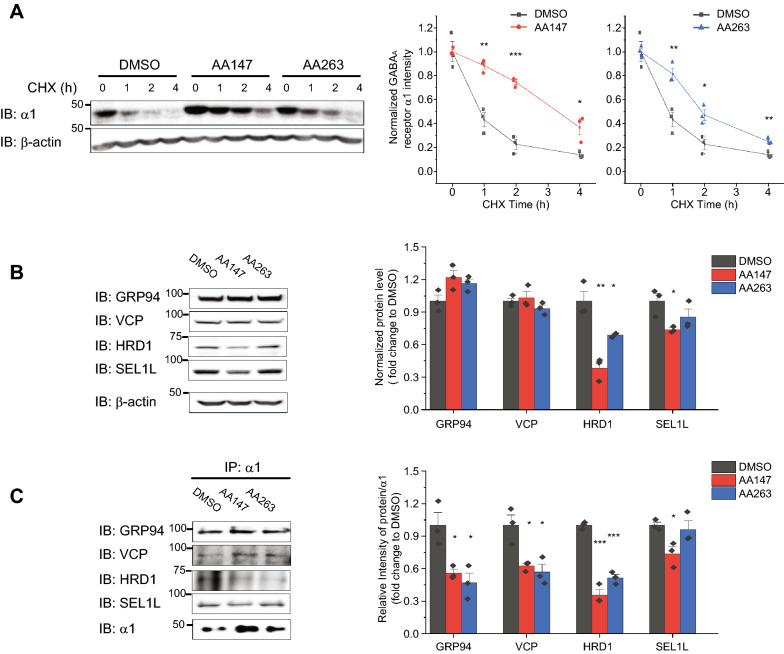
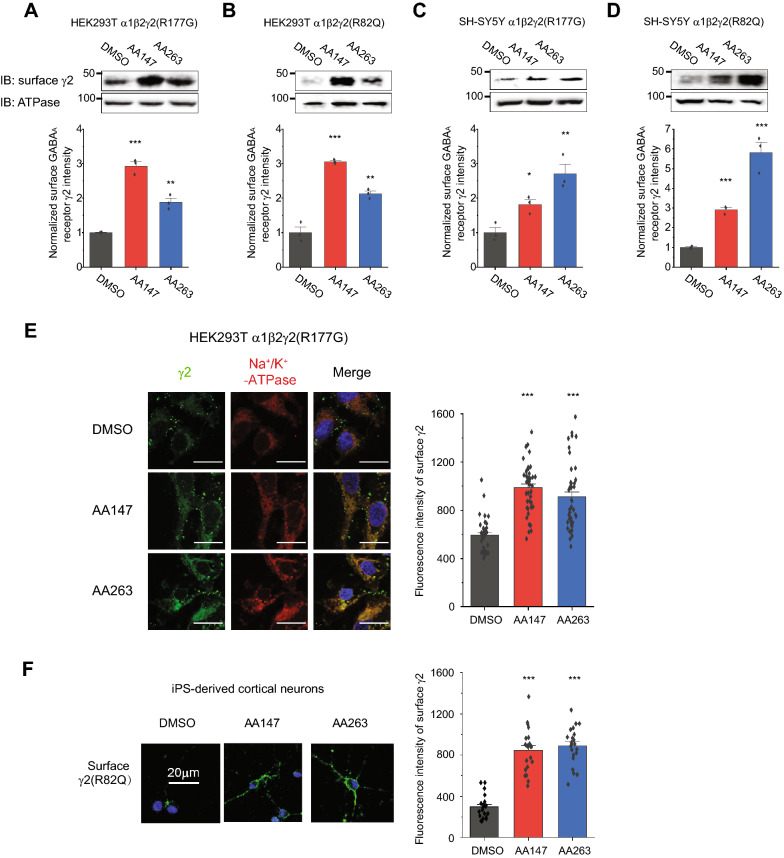
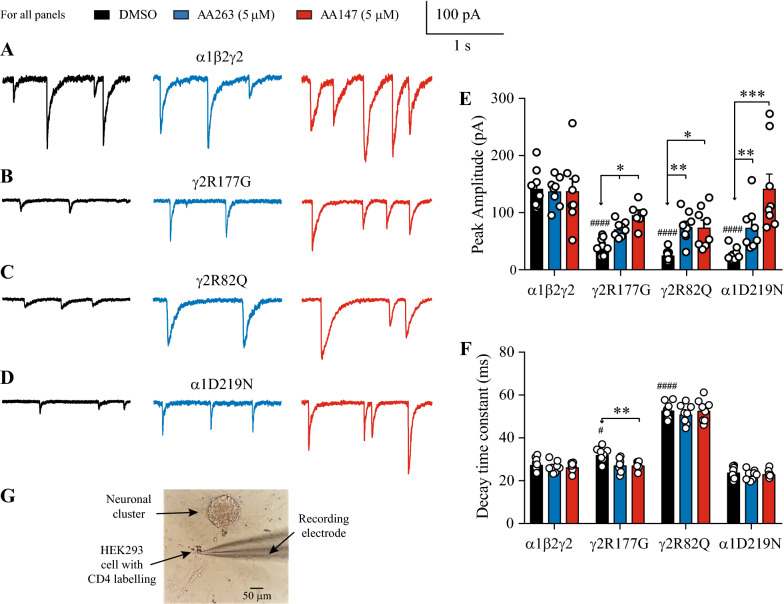
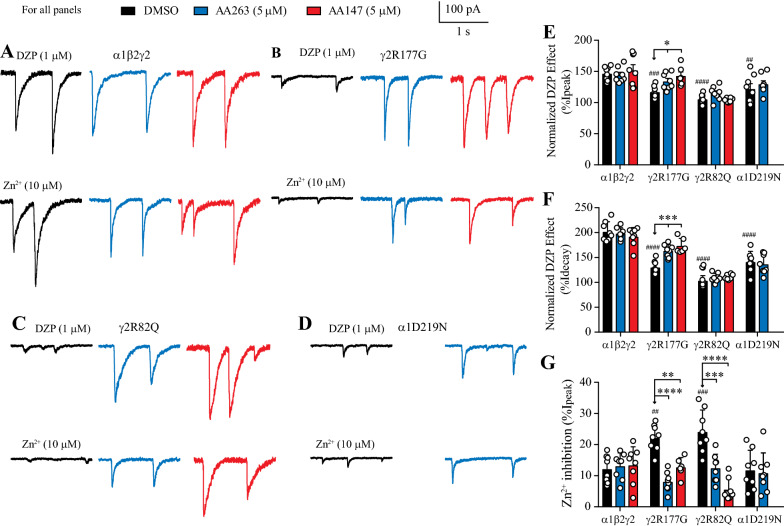
Here, we demonstrate that pharmacologically enhancing endoplasmic reticulum (ER) proteostasis using small molecule activators of the ATF6 (Activating Transcription Factor 6) signaling arm of the unfolded protein response (UPR) increases the assembly, trafficking, and surface expression of variant GABA receptors. These improvements are attributed to ATF6-dependent remodeling of the ER proteostasis environment, which increases protein levels of pro-folding ER proteostasis factors including the ER chaperone BiP (Immunoglobulin Binding Protein) and trafficking receptors, such as LMAN1 (Lectin Mannose-Binding 1) and enhances their interactions with GABA receptors. Importantly, we further show that pharmacologic ATF6 activators increase the activity of GABA receptors at the cell surface, revealing the potential for this strategy to restore receptor activity to levels that could mitigate disease pathogenesis.
These results indicate that pharmacologic ATF6 activators offer an opportunity to restore GABA receptor activity in diseases including genetic epilepsy and point to the potential for similar pharmacologic enhancement of ER proteostasis to improve trafficking of other disease-associated variant ion channels implicated in etiologically-diverse diseases.
γ-氨基丁酸A型(GABA)受体亚基的基因变异与包括遗传性癫痫在内的多种病理状况的发生有关。先前的研究表明,致病性GABA亚基会促进GABA受体的错误折叠和低效组装,限制受体在质膜上的表达和活性。然而,含有变异亚基的GABA受体仍可保留活性,这表明增强这些变异受体的折叠、组装和运输为减轻与遗传性癫痫相关的病理状况提供了潜在机会。
在此,我们证明,使用未折叠蛋白反应(UPR)的ATF6(激活转录因子6)信号臂的小分子激活剂从药理学上增强内质网(ER)蛋白质稳态,可增加变异GABA受体的组装、运输和表面表达。这些改善归因于ER蛋白质稳态环境的ATF6依赖性重塑,这增加了包括ER伴侣BiP(免疫球蛋白结合蛋白)和运输受体(如LMAN1,凝集素甘露糖结合蛋白1)在内的促折叠ER蛋白质稳态因子的蛋白质水平,并增强了它们与GABA受体的相互作用。重要的是,我们进一步表明,药理学上的ATF6激活剂可增加细胞表面GABA受体的活性,揭示了该策略将受体活性恢复到可减轻疾病发病机制水平的潜力。
这些结果表明,药理学上的ATF6激活剂为恢复包括遗传性癫痫在内的疾病中的GABA受体活性提供了机会,并指出了类似的药理学增强ER蛋白质稳态以改善其他与病因多样的疾病相关的变异离子通道运输的潜力。